Metabolomics DSP Course - Data Analysis#
In this demonstrative exercise, we will perform the downstream analysis of the Metabolights dataset MTBLS8735. This dataset contains three control samples derived from healthy samples, three samples from patients with Cardiovascular disease and four pooled Quality Control (QC) samples.
The rough workflow of this analysis is:
Data filtering
Imputation
Correcting instrumental drift
Normalisation and Visualisation
Statistical analysis (ANCOVA)
For most of the analyses, we will be using the python package ACORE, which is a package
developed by the Data Science Platform of NNF BRIGHT for analysing multi-omics molecular
data - including metabolomics data. The documentation of this package can be found here:
acore documentation. Go there to read
about the functions in more detail, or to find out what else you can analyse with acore.
Although we are using acore for almost everything, all of these analyses can be carried out with other programs or in your own code, created in R, Python or other places, as well. This notebook serves primarily as a demonstration of the necessary steps of a typical downstream metabolomics data analysis.
Imports#
import matplotlib.pyplot as plt
import matplotlib.transforms as transforms
import numpy as np
import pandas as pd
import vuecore.plots.basic.scatter
from matplotlib.patches import Ellipse
from sklearn.decomposition import PCA
from sklearn.preprocessing import StandardScaler
Helper functions#
First, we are defining some helper functions. Feel free to ignore these while we go through the exercise, they are for example plotting functions. Do, however, run the cells.
Data setup#
fname_features = "results_prepared/output_quantification_Linked_data.tsv"
fname_metadata = "data/MTBLS8735/metadata.csv"
First, let’s load and have a look at our data. You can find it in the data folder of this codespace.
metaboigniter_data = pd.read_csv(
fname_features,
sep="\t",
index_col=0,
dtype={"id": str},
)
metaboigniter_data
| charge | RT | mz | quality | MS_D_POS.mzML | MS_C_POS.mzML | MS_E_POS.mzML | MS_B_POS.mzML | MS_A_POS.mzML | MS_QC_POOL_1_POS.mzML | MS_QC_POOL_4_POS.mzML | MS_QC_POOL_3_POS.mzML | MS_F_POS.mzML | MS_QC_POOL_2_POS.mzML | adduct | feature_ids | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| id | ||||||||||||||||
| 1785900975374656749 | 0 | 82.991657 | 487.360409 | 0.000045 | 2197.7150 | 2499.92530 | 2055.4453 | 1789.1910 | 1737.8285 | 2228.8013 | 1863.83950 | 2093.6409 | 1761.39360 | 1882.0083 | NaN | [10439623260835028, 1044712869837677293, 82553... |
| 10359796054321139656 | 0 | 145.385393 | 141.958245 | 0.000026 | 1745.7393 | 987.70935 | 1540.5591 | 1862.1211 | 826.2672 | 555.4885 | 642.15070 | 1176.8750 | 997.84753 | 1006.0284 | [M+H]+ | [31491152637877450, 16826451592456168833, 8068... |
| 17114916068296246246 | 1 | 203.540336 | 151.035251 | 0.000984 | 43793.8500 | 42707.09000 | 44408.4000 | 35033.9800 | 47008.1200 | 31749.9700 | 31151.19000 | 33077.8100 | 42819.70000 | 33124.9000 | [M+H]+ | [121786566209455346, 16662184219603302854, 103... |
| 10875454315052802400 | 1 | 182.913495 | 342.015418 | 0.000209 | 14125.5400 | 11501.71000 | 9088.2600 | 6860.9673 | 12174.5300 | 1247.7310 | 2869.22780 | 2894.0437 | 12748.26000 | 4556.0920 | [M+H]+ | [130187692158764043, 8708957191126143269, 5655... |
| 15167360593113340842 | 1 | 111.636771 | 491.300119 | 0.004628 | 143542.6000 | 173214.20000 | 89948.0100 | 93468.9200 | 157576.2000 | 198321.3000 | 207862.40000 | 206423.1000 | 169903.50000 | 204664.5000 | [M+H]+ | [163715205785070798, 15732231754378009136, 181... |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| 17892945075043156858 | 0 | 125.446453 | 447.290728 | 0.000025 | 0.0000 | 0.00000 | 0.0000 | 0.0000 | 0.0000 | 0.0000 | 1229.67710 | 0.0000 | 0.00000 | 0.0000 | NaN | [18165040187458211252] |
| 2906889230784610727 | 0 | 33.937876 | 560.274926 | 0.000014 | 0.0000 | 0.00000 | 0.0000 | 0.0000 | 0.0000 | 0.0000 | 679.27590 | 0.0000 | 0.00000 | 0.0000 | NaN | [18167075448252607803] |
| 17135030643150808614 | 0 | 40.633406 | 341.263868 | 0.000013 | 0.0000 | 0.00000 | 0.0000 | 0.0000 | 0.0000 | 0.0000 | 659.15150 | 0.0000 | 0.00000 | 0.0000 | NaN | [18283503193542699509] |
| 2244705787916254982 | 0 | 33.937876 | 578.342658 | 0.000022 | 0.0000 | 0.00000 | 0.0000 | 0.0000 | 0.0000 | 0.0000 | 1072.48970 | 0.0000 | 0.00000 | 0.0000 | NaN | [18283940505064136015] |
| 10873393075126387345 | 0 | 35.611759 | 676.320660 | 0.000014 | 0.0000 | 0.00000 | 0.0000 | 0.0000 | 0.0000 | 0.0000 | 698.24835 | 0.0000 | 0.00000 | 0.0000 | NaN | [18291256843486362483] |
13094 rows × 16 columns
metaboigniter_data.columns
Index(['charge', 'RT', 'mz', 'quality', 'MS_D_POS.mzML', 'MS_C_POS.mzML',
'MS_E_POS.mzML', 'MS_B_POS.mzML', 'MS_A_POS.mzML',
'MS_QC_POOL_1_POS.mzML', 'MS_QC_POOL_4_POS.mzML',
'MS_QC_POOL_3_POS.mzML', 'MS_F_POS.mzML', 'MS_QC_POOL_2_POS.mzML',
'adduct', 'feature_ids'],
dtype='object')
You can see that we have our features in the rows, and that there are 9068 of them. We have some metadata like mass-to-charge ratios and retention times, and then we have our intensities.
In order to properly analyse our data, we have to transpose it first. We also have to remove the metadata columns.
data = metaboigniter_data.T
data = data.drop(["charge", "RT", "mz", "quality", "adduct", "feature_ids"])
data = data.where(data > 1, np.nan)
data
| id | 1785900975374656749 | 10359796054321139656 | 17114916068296246246 | 10875454315052802400 | 15167360593113340842 | 7744533318583132094 | 1718920057707733684 | 6744901733644451055 | 11872048905895506999 | 13329452032929808115 | ... | 3029767587063264539 | 17167452654274772591 | 8248272264971643600 | 3480066978786852155 | 17040961437395340439 | 17892945075043156858 | 2906889230784610727 | 17135030643150808614 | 2244705787916254982 | 10873393075126387345 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MS_D_POS.mzML | 2197.715 | 1745.7393 | 43793.85 | 14125.54 | 143542.6 | 1585.027 | 7369.8022 | 5886.109 | 11476.87 | 3449.8782 | ... | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN |
| MS_C_POS.mzML | 2499.9253 | 987.70935 | 42707.09 | 11501.71 | 173214.2 | 1210.8177 | 5918.5693 | 4802.169 | 10514.19 | 2679.616 | ... | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN |
| MS_E_POS.mzML | 2055.4453 | 1540.5591 | 44408.4 | 9088.26 | 89948.01 | 4690.5874 | 6250.251 | 5141.211 | 10575.09 | 2356.0645 | ... | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN |
| MS_B_POS.mzML | 1789.191 | 1862.1211 | 35033.98 | 6860.9673 | 93468.92 | 5135.8164 | 6641.842 | 2480.7393 | 8486.273 | 3797.3372 | ... | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN |
| MS_A_POS.mzML | 1737.8285 | 826.2672 | 47008.12 | 12174.53 | 157576.2 | 2573.8994 | 5735.305 | 6731.2915 | 9867.111 | 6096.421 | ... | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN |
| MS_QC_POOL_1_POS.mzML | 2228.8013 | 555.4885 | 31749.97 | 1247.731 | 198321.3 | 8646.111 | 8258.921 | 4336.047 | 9133.133 | 4147.0293 | ... | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN |
| MS_QC_POOL_4_POS.mzML | 1863.8395 | 642.1507 | 31151.19 | 2869.2278 | 207862.4 | 6730.96 | 9196.043 | 4037.6926 | 9277.422 | 4504.331 | ... | 666.54095 | 2721.9639 | 1475.8854 | 954.4274 | 1300.5066 | 1229.6771 | 679.2759 | 659.1515 | 1072.4897 | 698.24835 |
| MS_QC_POOL_3_POS.mzML | 2093.6409 | 1176.875 | 33077.81 | 2894.0437 | 206423.1 | 8941.185 | 9147.992 | 4297.844 | 9498.344 | 5428.016 | ... | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN |
| MS_F_POS.mzML | 1761.3936 | 997.84753 | 42819.7 | 12748.26 | 169903.5 | 2100.4517 | 6168.7725 | 5676.127 | 10521.12 | 2820.938 | ... | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN |
| MS_QC_POOL_2_POS.mzML | 1882.0083 | 1006.0284 | 33124.9 | 4556.092 | 204664.5 | 7594.9097 | 7804.082 | 4517.161 | 9515.199 | 4617.7646 | ... | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN | NaN |
10 rows × 13094 columns
Now our data consists of only features in the columns, and samples in the rows.
We are also going to define some variables that help us later on in the code.
samples = [
"MS_F_POS.mzML",
"MS_E_POS.mzML",
"MS_C_POS.mzML",
"MS_A_POS.mzML",
"MS_B_POS.mzML",
"MS_D_POS.mzML",
]
samples_cvd = ["MS_F_POS.mzML", "MS_A_POS.mzML", "MS_D_POS.mzML"]
samples_ctr = ["MS_E_POS.mzML", "MS_C_POS.mzML", "MS_B_POS.mzML"]
qcs = [
"MS_QC_POOL_1_POS.mzML",
"MS_QC_POOL_2_POS.mzML",
"MS_QC_POOL_3_POS.mzML",
"MS_QC_POOL_4_POS.mzML",
]
groups_samples = {
"CTR": samples_ctr,
"CVD": samples_cvd,
}
groups_all = {
"CTR": samples_ctr,
"CVD": samples_cvd,
"QC": qcs,
}
Data filtering#
As a first step, we need to filter our data. There are many features in the data which are artifacts, background noise or unreliable. Additionally, there is a lot of missingness. While missing values can be imputed, it’s better to remove features with high levels of missingness first and only impute features that have signal in most samples.
We will use three methods of filtering:
80%-rule
Coefficient of Variation (CV)-based filtering
Dispersion ratio (D-ratio) filtering
For most of these, we will use the filter_metabolomics module from acore.
from acore import filter_metabolomics as fm
Let’s first check how much missingness there is in the data.
data.isna().sum().sum()
missing_per_feature = data.isna().sum(axis=0)
missing_per_feature
freq_table = missing_per_feature.value_counts().sort_index()
df_freq = freq_table.reset_index()
df_freq.columns = ["n_missing", "n_features"]
df_freq
| n_missing | n_features | |
|---|---|---|
| 0 | 0 | 1,084 |
| 1 | 1 | 369 |
| 2 | 2 | 314 |
| 3 | 3 | 383 |
| 4 | 4 | 491 |
| 5 | 5 | 577 |
| 6 | 6 | 985 |
| 7 | 7 | 1,106 |
| 8 | 8 | 1,801 |
| 9 | 9 | 5,984 |
plot_feature_missingness(data)
plot_intensity_distribution(data)
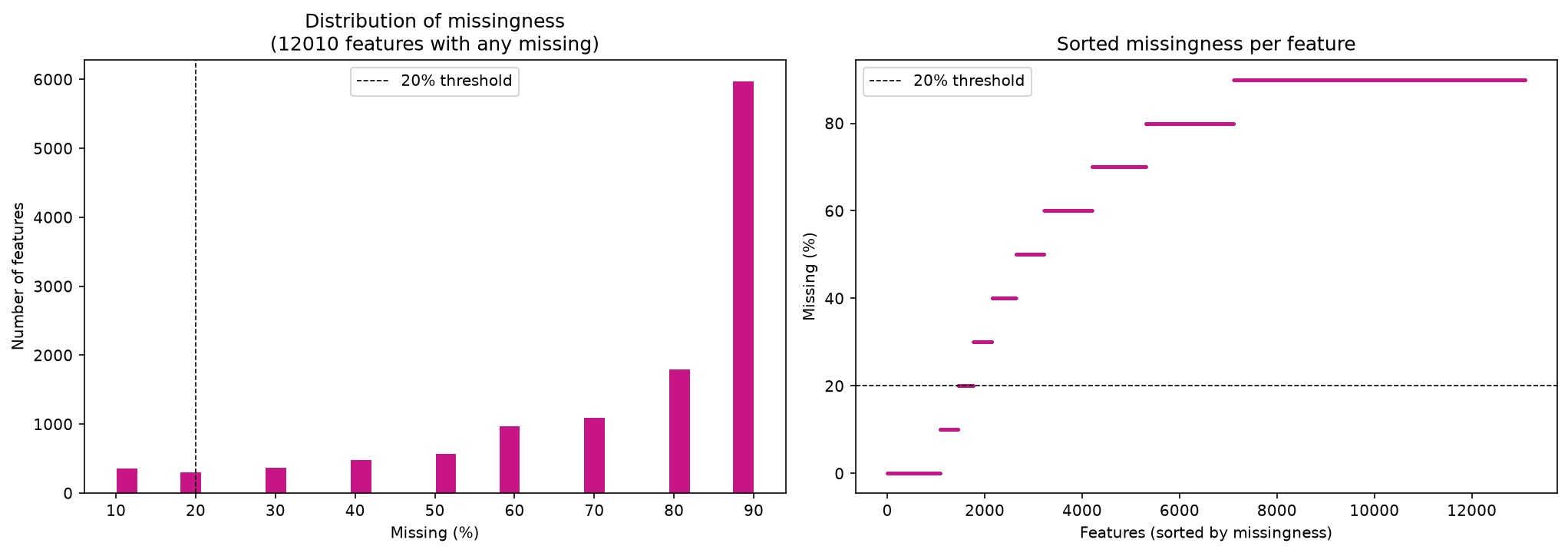
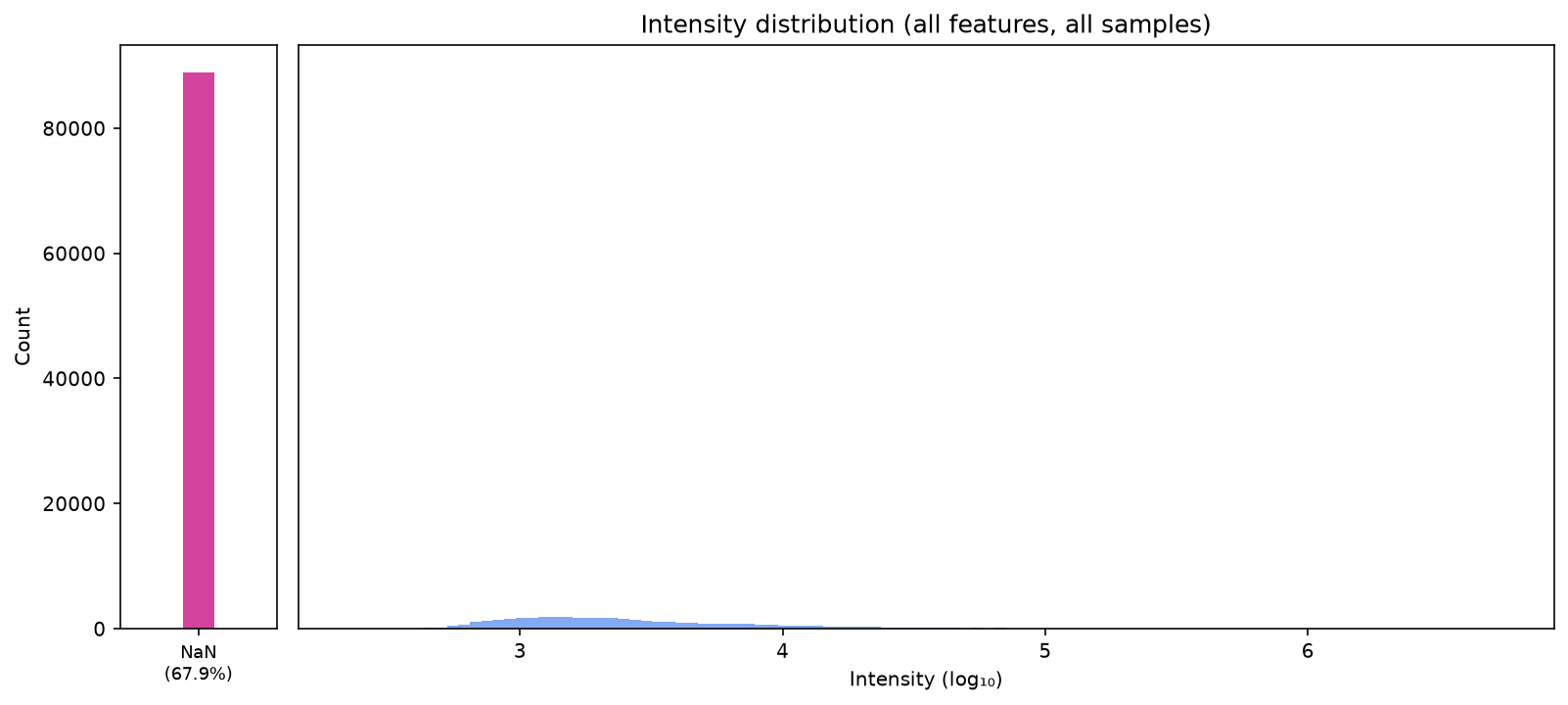
There are a lot of missing values, so it is important that we filter first.
80%-rule for filtering#
The 80%-rule filters out features with too much missingness from our data. More specifically, if for a feature, more than 20% of the data is missing across all sample columns, it will be removed, so features must have at least 80% of data present in order to be retained.
Although it is called the 80%-rule, other thresholds can be used to make the filtering more lenient or more stringent.
In acore, this method is implemented in the function filter_by_missingness. Let’s first have a look at our function.
help(fm.filter_by_missingness)
Help on function filter_by_missingness in module acore.filter_metabolomics:
filter_by_missingness(data: pandas.core.frame.DataFrame, percent: int = 80, method: str = 'classic', samples: list | None = None, groups: dict | str | None = None)
Implementation of the 80%-rule.
If there are more than 20% of values (intensities) missing for one feature,
this feature will get removed.
:param data: pandas data frame with samples as rows and features as columns.
:param percent: percentage chosen for filtering. The default is 80%, meaning that
at least 80% of the values of every feature need to be present in order for this
feature to be retained.
:param method: str that is either "classic" or "modified".
If "classic", all samples are considered for each feature. Samples are taken from the
"samples" parameter and should not include controls or QCs.
If "modified", conditions are separated when calculating the percentage of
missingness. A feature is retained if at least ``percent``% of its values are
present in ANY one condition. This allows condition-specific features (e.g.
present in treatment but missing in control) to be retained.
:param samples: list of row index labels (from data.index) identifying the biological
sample rows, e.g. ["S1", "S2", "S3"]. Required when method="classic". Should not
include control or QC samples.
:param groups: required when method="modified", ignored otherwise. Can be either:
- A dict mapping condition name to a list of row index labels belonging to that
condition, e.g. ``{"treatment": ["S1", "S2", "S3"], "control": ["S4", "S5"]}``.
QCs and blanks are excluded by simply not including them in the dict.
- A str naming a column in ``data`` whose values define the condition for each row,
e.g. ``"sample collection"`` if rows carry values like ``"Berlin"``, ``"Copenhagen"``, ``"London"``.
Every unique value in that column becomes a condition group containing all rows
with that value. When using this option, make sure to not include any other metadata
columns in the data frame.
data_filtered_1 = fm.filter_by_missingness(
data=data,
method="classic",
percent=80, # 80% present, 20% missing is allowed at most
samples=samples,
)
print(
f"Num. of features before filtering: {data.shape[1]}\n"
f"Num. of features after filtering: {data_filtered_1.shape[1]}"
)
print(f"Difference: {data.shape[1]-data_filtered_1.shape[1]} features removed.")
Num. of features before filtering: 13094
Num. of features after filtering: 2030
Difference: 11064 features removed.
missing_per_feature = data_filtered_1.isna().sum(axis=0)
freq_table = missing_per_feature.value_counts().sort_index()
df_freq = freq_table.reset_index()
df_freq.columns = ["n_missing", "n_features"]
df_freq
| n_missing | n_features | |
|---|---|---|
| 0 | 0 | 1,084 |
| 1 | 1 | 369 |
| 2 | 2 | 156 |
| 3 | 3 | 143 |
| 4 | 4 | 161 |
| 5 | 5 | 117 |
Now we have removed a huge number of features, and we have 2402 features left, which is a number we can trust more to contain real metabolites.
CV-based filtering#
In this method, we are taking into account the quality control (QC) samples.
The CV of the biological samples and the CV of the QC samples are calculated per feature, and if for a given feature the CV of the QC samples is larger than that of the biological samples, it is removed. Also, if there are not enough QC samples to calculate the CV, or the mean is near zero, the features will be removed.
In acore, this method is implemented in the function filter_cv.
data_filtered_2 = fm.filter_cv(data=data_filtered_1, samples=samples, qcs=qcs)
CV is undefined for 354 feature(s) in QC samples (zero or near-zero mean): ['2636573287799528368', '1255015684874353805', '2081128107789029548', '5781440246377970407', '4348073021217059041', '13220515123801564329', '5735706107592489893', '15507882067355905462', '15119977206972289711', '3362025122796601167', '3913658956857853394', '8387958378096062392', '3819034200272550136', '4870725809536008143', '3368940459997904894', '511158606928777651', '8359779065487926943', '17030460597289483924', '4137860821822396914', '8185010247011980449', '6771822454118994987', '15723084508438160594', '14470900064776210944', '16914230235921142351', '6553421105201890147', '4450470613056901292', '4333167807014612237', '10093127670235868684', '14788295203148016883', '8225391745561756962', '7698939552977997768', '17080061998131909992', '10200907477884162507', '9574464210701828047', '8630550943888457114', '11357582629299129612', '9716372561691699674', '12581297789861346000', '16324985885956160892', '10624439122852148279', '10404019867363800846', '361914675579226375', '16398547070634316985', '3916914144309896527', '5668830318206055822', '16967037721730340430', '509634750750314099', '3570994927278375199', '781152180314057369', '5035024713025228949', '13031927439947541059', '14501409465138175694', '15377367450991565584', '9903459879052056188', '10147781995224059332', '7052353277864808363', '5312557798985953381', '2251945460469300124', '8474372732815718116', '4931818636333551622', '13294741869978890633', '14037926341379447302', '16837613254721991893', '16595686959354083926', '12305003632565459877', '4610803311769876877', '3621968061231047547', '18011491880988591294', '11963766406844959833', '14954585624193348775', '5663416294484494586', '6913368617372373913', '7280347929962952086', '17071282131790106652', '12144752853475610333', '15077286983107585926', '4304792257240652264', '14324159019850352288', '7929278616402482928', '10796132678596579388', '15189011702119082443', '16279993413619078309', '6309562004239164489', '5804270525674648462', '798289847853653743', '15000975580483540155', '13656002642858725142', '2964868095064882997', '14769065035125174322', '11565401818044424100', '3431462258295024329', '15296182729037581803', '12059195505431450398', '16483697613972222617', '1995522595466210123', '13829051245229107055', '2367774217195777392', '4573185524527043559', '10818236428169971352', '5841097871547350073', '2623152618911033112', '12843468090252313891', '10005728109943372585', '11363824999851726797', '13696550288222535919', '14813702858422464516', '10148778884392650630', '6040984414437521626', '569844582994903810', '15922918882973105274', '8848030522974059219', '14977245175216057173', '10898674941812780916', '15040933028001137811', '16618575542935013112', '13378297485658227745', '5120777930797758415', '2245942744886786316', '13668648066553381335', '15370752943426116923', '17775728625728526300', '15240823336139229434', '7591070574604691042', '16085717739781018295', '11702243547092109217', '14294418627620382612', '5145409808297484753', '504021836781857864', '852743475633300514', '11797778866067310609', '15173772532583121018', '1165337272744792852', '8541474378159698633', '10209798891472981577', '6814339247632592482', '9873230281782298625', '5676514744616969634', '13844132613006394423', '4496034635078497417', '17476295575867517193', '16931641582785208643', '3315879149328782926', '11205886417941204219', '15845550851592783049', '786129637942103822', '6666602386750039935', '1168409256533927296', '12467458281524608346', '2016133159332726490', '872574529216161932', '7638185182949747120', '15852436465056532869', '4999089546644570558', '3830678336944555965', '8447010120403248848', '2544376217953093795', '10228659080742513111', '9790404852889486271', '16920394540702587106', '3046892850389242532', '3279185111149384169', '4275502339032208967', '18139568930188156507', '6234624610047017104', '1435275024794032570', '9652393893165833881', '10050357070071599350', '6656450922543626022', '606504257672388797', '13911299966387436413', '10548317586042806642', '16926395594954367023', '13837383007057849235', '1743482886289977573', '10326568740122203040', '14100931090787203178', '15490081426871572512', '18172164892415525395', '4952425199812453011', '2310936618461611468', '13351055964946043480', '3638366590243975347', '17173627733445787208', '11724155391674287797', '7324333339247511249', '3724158625030667196', '3391818906289683450', '5755226549709800323', '6309354348887081983', '14954049772349002116', '16467792741850615819', '3084778339629661693', '11229823620508563093', '6952400408038480831', '13771814307344361242', '8932690292948955', '8346553042974637769', '16374387492440682455', '2321734149534721747', '13102179682146098023', '9084647485922047589', '17868889705766239494', '13411107527625914178', '6867978268651814809', '5054880329892884241', '564408538531169013', '2589825494090942625', '15021706807689077259', '1430565569916702968', '6136261705834320425', '16754674896460313250', '4638973274841339587', '11306761391520723871', '8663630229512504449', '14592975655118886795', '13255758580880342862', '5783010915073843821', '2140443717645846237', '15068017531177497683', '2771791507981697852', '11938149362346260298', '13632889303241824010', '221282040873628381', '2360116252060786144', '557447042648650761', '10599166198379912823', '5268176105357147441', '853762737751922241', '12874001305712922813', '9615730657356806643', '3844766510055785390', '2160572170955076417', '12219490980155197564', '2857950114216426228', '12474737476957198329', '16852993381434846063', '13014825145373798647', '10728269715318340025', '17786663879971390761', '8642504770045222449', '1204300380755777468', '633408373817680023', '6880932652492261920', '3722608177106844010', '16184974079729046694', '1939254642554869161', '292789874130935080', '14742478120963365923', '16383419911473284996', '3895273031840824416', '12739127595742812487', '13623807241875171107', '14814331162944008281', '18218726121609328943', '8700350614288732135', '4802062320480388343', '10552173463747256886', '3715948001047078886', '4104508007368224884', '14960753641169051895', '3269563522786161920', '6490605393381218772', '6429742392879766599', '8022606716345410307', '18016209946099875690', '2016690505670333723', '7188636343372679305', '7985568031692605185', '7634625683811627794', '16605864908334800945', '75928148421214418', '7113737177577096169', '12862042232485741910', '555810434558088652', '17972190719759214645', '14867484644649144816', '1823280901220429830', '10110894258813285795', '15261578288174232896', '12648589632450305283', '942147640863031077', '10119650417219737610', '16966311955077689846', '12680383144001528552', '18030652240498986760', '5520129959690665178', '6831368997084644248', '11828710142511208046', '13275918157615948605', '8712780164805136735', '8912077449492896368', '15908028234949193679', '6266312799824645107', '2053467111543157513', '17688481739501524301', '15144513882473912555', '4514957885424491649', '16885518096602862573', '9454377282322109999', '4984585598939123929', '17593396409995702123', '6534808621076036599', '8626849701027317034', '12665414741454062749', '11643549461492340283', '3125052634771617587', '8615106625112788855', '8269573480153202630', '199578909385456580', '16416278511861675207', '17081164027085617701', '12959759930992894854', '13175829387719373675', '7355225553120660872', '6843905031274338767', '13139392019073770412', '17452197076971765207', '4427264037567165339', '46737384901994729', '12369362647558541692', '1886883562090945349', '13266220471282669796', '4602051368890405189', '12984543218258452595', '4274087984323434198', '5109497063857380894', '14875143583591801393', '5757588107067960822', '11758984089835601860', '2375118766094095626', '50529216970010884', '2261199985994379179', '7974993312250601020', '187711411102438322', '1314667531947192054', '7275293278521561801', '2720939922466084551', '12428580384977482891', '15504276382164479158', '7324000700810999245', '4444732873093030575', '2399938284904877714', '14584580378454801206', '16025334128187851531', '12349149626282790755', '2990490906593941946', '10461853759643938022', '1247510545602223557', '5953143397292869381', '8628406943857354296', '1715494497952354133', '6978328347033344613', '13490971593716844149', '2718471385430446022']. These features will be dropped. Consider running filter_by_missingness first.
print(
f"Num. of features before filtering: {data_filtered_1.shape[1]}\n"
f"Num. of features after filtering: {data_filtered_2.shape[1]}"
)
print(
f"Difference: {data_filtered_1.shape[1]-data_filtered_2.shape[1]} features removed."
)
Num. of features before filtering: 2030
Num. of features after filtering: 1416
Difference: 614 features removed.
D-ratio filtering#
Finally, we will do dispersion ratio filtering.
This method scores each feature by the ratio of its analytical noise (QC) to its total variation, meaning the standard deviation across pooled QC injections divided by the standard deviation across biological samples.
A low D-ratio means biological variation dominates the technical noise, and a high one means the feature is mostly measurement noise with little biological signal, so features above a threshold (typically 0.5 or lower) get removed.
Here, we will use the median absolute deviation (MAD, scaled by 1.4826). Because medians ignore extreme values, this version resists outliers and skew, making it the better choice for non-Gaussian data like raw untargeted peak areas — whereas the standard-deviation version is preferable when the data are roughly Gaussian (or have been log-transformed first).
This method is not implemented in acore yet, so we will define it ourselves.
def filter_dratio(
data: pd.DataFrame,
samples: list,
qcs: list,
threshold: float = 0.5,
):
if len(qcs) < 2:
raise ValueError(
f"You need more than 1 QC sample to apply this filtering method. Got {len(qcs)}."
)
if len(samples) < 2:
raise ValueError(
f"You need more than 1 biological sample to apply this filtering method. "
f"Got {len(samples)}."
)
def mad(df): # median absolute deviation
# 1.4826 * median(|x - median(x)|), an unbiased SD estimate for Gaussian data.
med = df.median()
return 1.4826 * (df - med).abs().median()
df = data.copy()
disp_qcs = mad(df.loc[qcs])
disp_samples = mad(df.loc[samples])
dratio = disp_qcs / disp_samples
undefined = dratio.isna() | dratio.isin([np.inf, -np.inf])
if undefined.any():
print(
f"D-ratio is undefined for {undefined.sum()} feature(s) (zero or near-zero biological "
f"dispersion): {list(df.columns[undefined])}. These features will be dropped.",
)
keep = (dratio <= threshold) & ~undefined
return df.loc[:, keep]
data_filtered_3 = filter_dratio(
data=data_filtered_2, samples=samples, qcs=qcs, threshold=0.4
)
print(
f"Num. of features before filtering: {data_filtered_2.shape[1]}\n"
f"Num. of features after filtering: {data_filtered_3.shape[1]}"
)
print(
f"Difference: {data_filtered_2.shape[1]-data_filtered_3.shape[1]} features removed."
)
Num. of features before filtering: 1416
Num. of features after filtering: 897
Difference: 519 features removed.
print(f"Now we have a total of {data_filtered_3.shape[1]} features left.")
Now we have a total of 897 features left.
data_filtered_3.isna().sum().sum()
missing_per_feature = data_filtered_3.isna().sum(axis=0)
missing_per_feature
freq_table = missing_per_feature.value_counts().sort_index()
df_freq = freq_table.reset_index()
df_freq.columns = ["n_missing", "n_features"]
df_freq
| n_missing | n_features | |
|---|---|---|
| 0 | 0 | 610 |
| 1 | 1 | 176 |
| 2 | 2 | 82 |
| 3 | 3 | 29 |
Imputation#
While we have filtered out a lot of missingness, we still have missing values in our data. Those are filled in with imputation. Imputation methods for metabolomics are also implemented in acore. Here we will use the half minimum to impute our values with.
from acore.imputation_analysis import (
imputation_half_minimum,
imputation_zeros,
)
But first, let’s re-assess how much we need to fill in.
print(f"Total count of missing cells: {data_filtered_3.isnull().sum().sum()}")
print(
f"Overall percentage of missingness: {data_filtered_3.isnull().mean().mean() * 100}\n"
)
plot_feature_missingness(data_filtered_3)
plot_intensity_distribution(data_filtered_3)
Total count of missing cells: 427
Overall percentage of missingness: 4.760312151616499
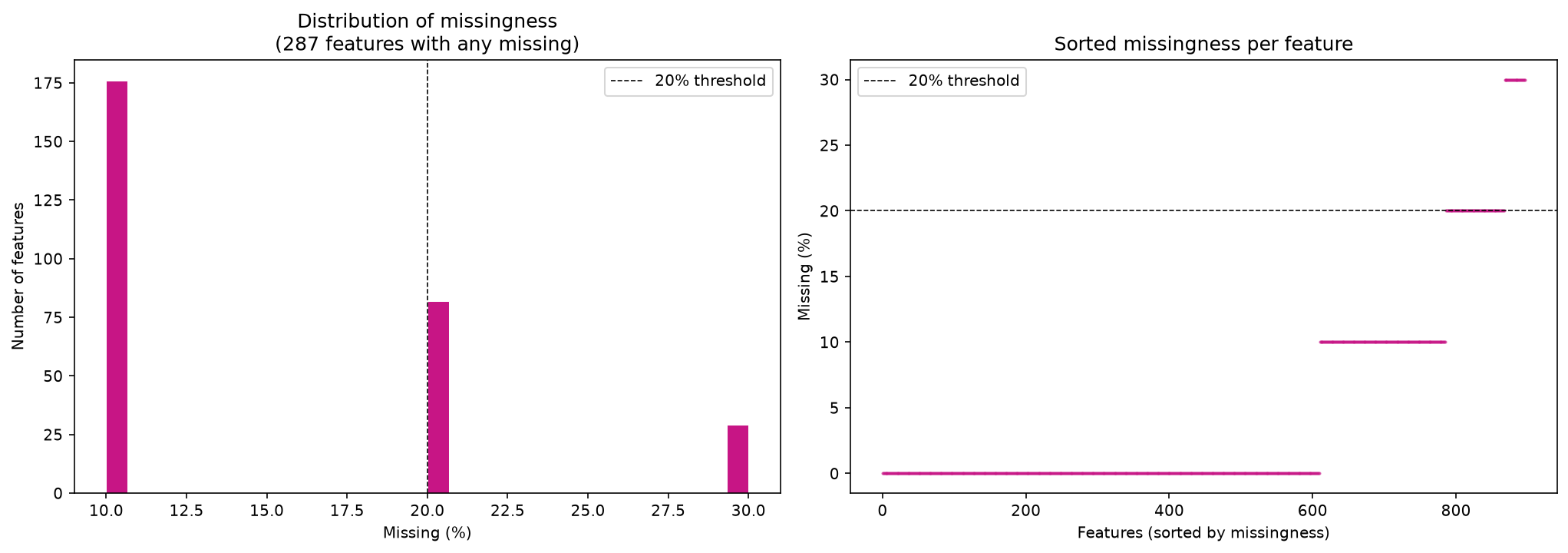
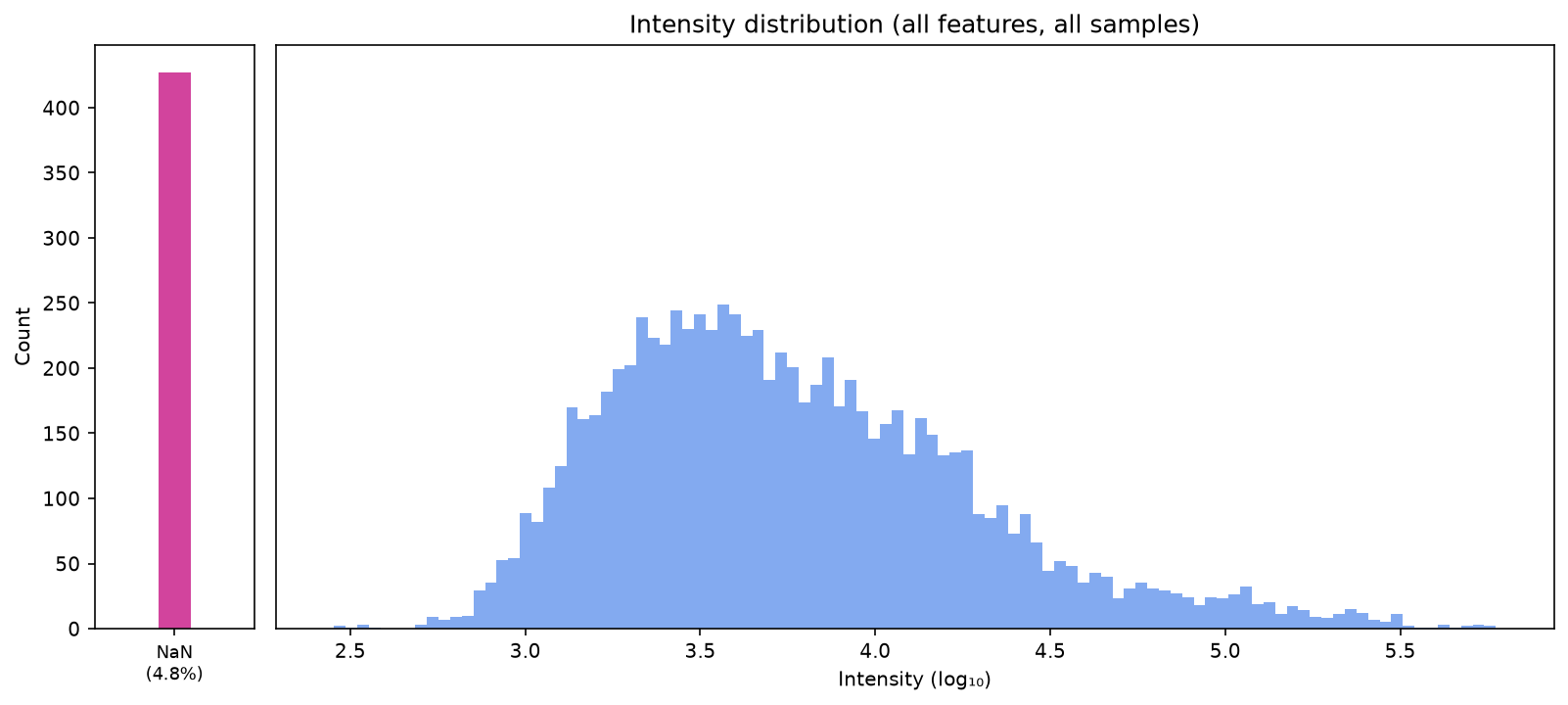
Impute with half minimum#
In this imputation method, we are calculating the minimum of each feature, taking the half of that and using that value to fill in missing values for that feature. This method is implemented in the acore function imputation_half_minimum().
Note: Another commonly used method in metabolomics data imputation is imputing with zeros. This method can also be applied through acore, with the function imputation_zeros().
data_imputed = imputation_half_minimum(data=data_filtered_3)
data_imputed
| id | 15167360593113340842 | 6744901733644451055 | 11872048905895506999 | 17709464339328039509 | 13922238524316478464 | 7261731678087078534 | 5009880893163685891 | 14568390051812142800 | 12965064019618390832 | 12018024353057911182 | ... | 10081236272997946842 | 11566491653082879729 | 1138090438525564760 | 16421084926908170456 | 15756805090958850402 | 6914550508003274460 | 8218613456253687867 | 1747965644941981794 | 8361543175644692425 | 9120899724421101875 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MS_D_POS.mzML | 143,542.600 | 5,886.109 | 11,476.870 | 20,796.740 | 12,335.740 | 1,841.792 | 13,084.690 | 65,376.190 | 46,063.120 | 6,191.987 | ... | 3,473.264 | 274.633 | 2,858.896 | 2,421.536 | 1,222.554 | 952.219 | 5,434.947 | 15,157.630 | 2,055.739 | 3,006.401 |
| MS_C_POS.mzML | 173,214.200 | 4,802.169 | 10,514.190 | 18,960.890 | 16,236.390 | 2,675.053 | 13,589.740 | 71,279.240 | 18,524.640 | 5,931.635 | ... | 9,403.579 | 704.575 | 1,666.042 | 858.752 | 1,444.158 | 1,444.071 | 5,278.917 | 14,413.630 | 1,598.920 | 1,444.313 |
| MS_E_POS.mzML | 89,948.010 | 5,141.211 | 10,575.090 | 21,024.620 | 17,866.430 | 2,610.558 | 12,433.400 | 52,452.010 | 59,626.340 | 5,618.113 | ... | 13,407.410 | 1,113.405 | 5,174.449 | 2,836.493 | 2,593.458 | 1,847.802 | 3,992.441 | 15,820.330 | 1,789.678 | 1,991.497 |
| MS_B_POS.mzML | 93,468.920 | 2,480.739 | 8,486.273 | 21,715.490 | 11,622.170 | 4,003.669 | 12,023.290 | 61,131.850 | 69,804.600 | 5,421.544 | ... | 10,555.010 | 888.467 | 3,474.066 | 3,253.217 | 2,190.118 | 1,100.630 | 4,106.935 | 15,740.730 | 4,263.249 | 1,911.875 |
| MS_A_POS.mzML | 157,576.200 | 6,731.292 | 9,867.111 | 19,383.730 | 19,050.570 | 2,090.373 | 12,986.100 | 65,314.800 | 53,463.480 | 5,762.296 | ... | 9,976.974 | 615.012 | 3,445.868 | 2,292.947 | 611.277 | 476.110 | 832.952 | 7,206.815 | 799.460 | 722.156 |
| MS_QC_POOL_1_POS.mzML | 198,321.300 | 4,336.047 | 9,133.133 | 23,995.310 | 9,895.060 | 2,919.634 | 10,952.520 | 54,107.960 | 31,009.850 | 5,038.122 | ... | 3,473.264 | 745.005 | 833.021 | 2,352.086 | 1,371.443 | 476.110 | 1,773.438 | 15,014.210 | 799.460 | 2,316.048 |
| MS_QC_POOL_4_POS.mzML | 207,862.400 | 4,037.693 | 9,277.422 | 23,487.570 | 11,016.590 | 3,455.879 | 11,468.140 | 58,678.800 | 36,071.060 | 5,318.160 | ... | 6,989.383 | 724.883 | 4,274.623 | 858.752 | 1,310.173 | 1,025.298 | 832.952 | 14,801.370 | 2,531.686 | 722.156 |
| MS_QC_POOL_3_POS.mzML | 206,423.100 | 4,297.844 | 9,498.344 | 24,143.250 | 10,479.450 | 3,532.990 | 11,710.650 | 56,706.770 | 32,887.300 | 5,395.765 | ... | 6,946.528 | 274.633 | 833.021 | 2,311.549 | 611.277 | 476.110 | 832.952 | 7,206.815 | 2,659.393 | 2,306.081 |
| MS_F_POS.mzML | 169,903.500 | 5,676.127 | 10,521.120 | 19,712.100 | 23,974.610 | 1,996.034 | 10,025.950 | 66,881.700 | 25,845.320 | 4,456.026 | ... | 9,844.492 | 549.266 | 833.021 | 1,717.504 | 1,296.626 | 1,237.158 | 5,141.985 | 15,161.050 | 1,860.046 | 1,595.857 |
| MS_QC_POOL_2_POS.mzML | 204,664.500 | 4,517.161 | 9,515.199 | 23,829.240 | 12,349.160 | 3,553.620 | 11,306.920 | 58,920.040 | 32,103.840 | 5,273.946 | ... | 3,473.264 | 274.633 | 4,513.373 | 858.752 | 611.277 | 1,068.735 | 1,665.903 | 7,206.815 | 799.460 | 722.156 |
10 rows × 897 columns
print(f"Total count of missing cells: {data_imputed.isnull().sum().sum()}")
print(
f"Overall percentage of missingness: {data_imputed.isnull().mean().mean() * 100}\n"
)
print("SUMMARY of imputation changes:")
missingness_summary(data, data_imputed)
plot_intensity_distribution(data_imputed)
Total count of missing cells: 0
Overall percentage of missingness: 0.0
SUMMARY of imputation changes:
Total values : 130,940
Missing before : 88,911 (67.9%)
Missing after : 0 (0.0%)
Features affected : 12010 / 13094
Samples affected : 10 / 10
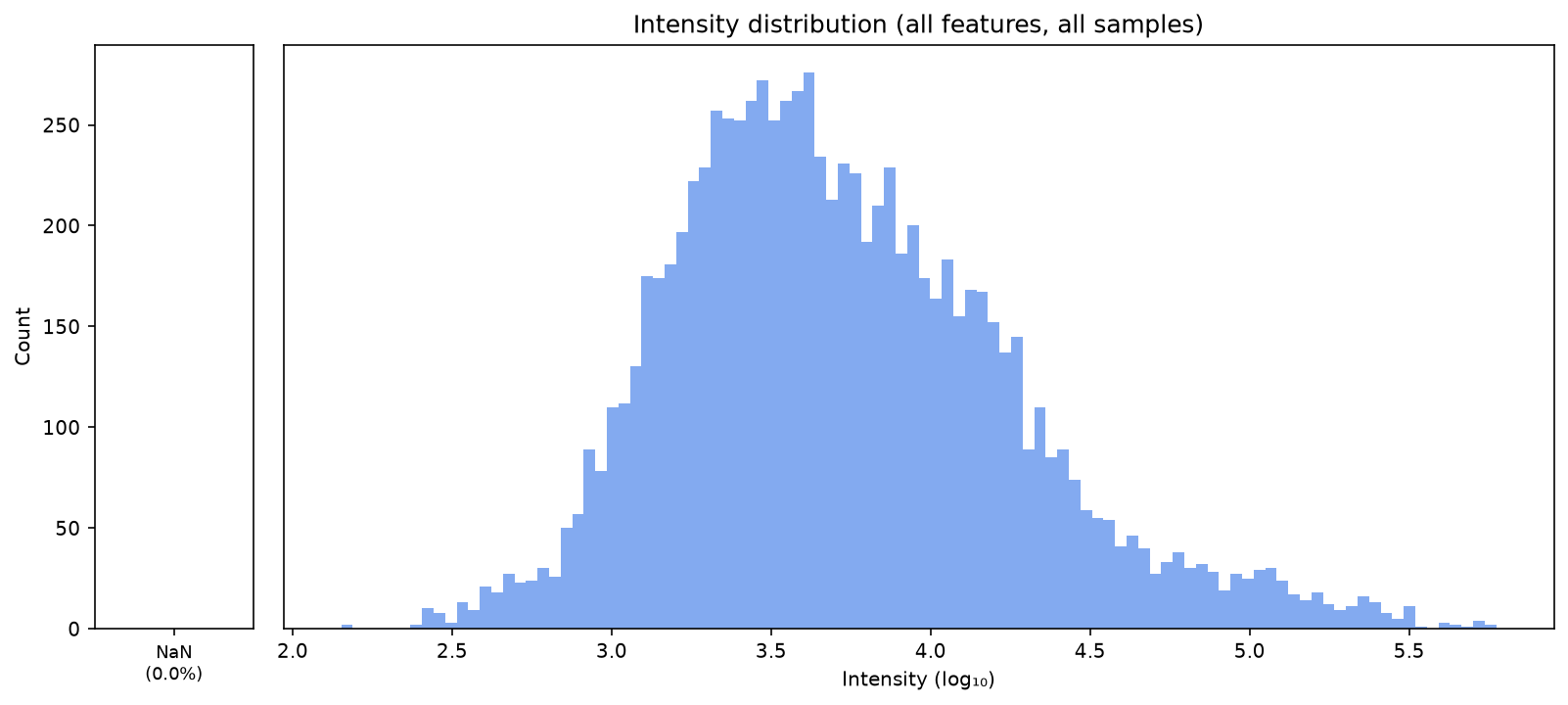
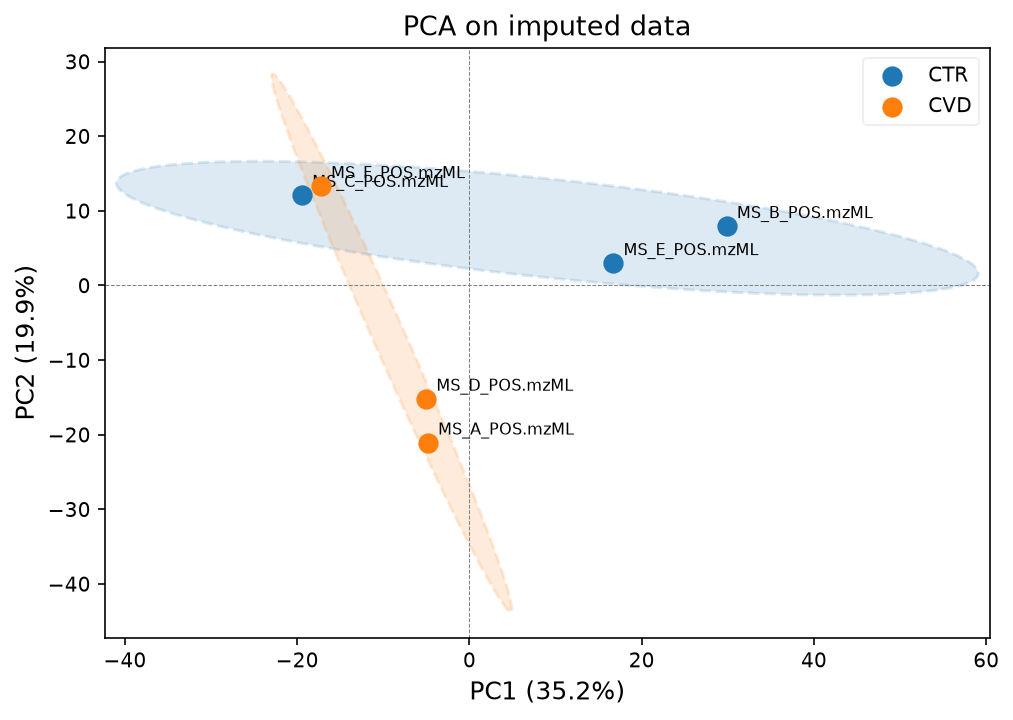
Now we have filled in all missing values. We can now plot the data in a PCA plot to see whether the samples can be separated well with principal components. We can use the functions we defined earlier at the beginning of the notebook for that.
We can plot the PCA first with all samples and QCs, and then with just the samples, to see how the groups separate.
pca_model, scores, var_explained = plot_pca(
data_imputed, groups_all, label_points=True, title="PCA on imputed data"
)
pca_model, scores, var_explained = plot_pca(
data_imputed, groups_samples, label_points=True, title="PCA on imputed data"
)
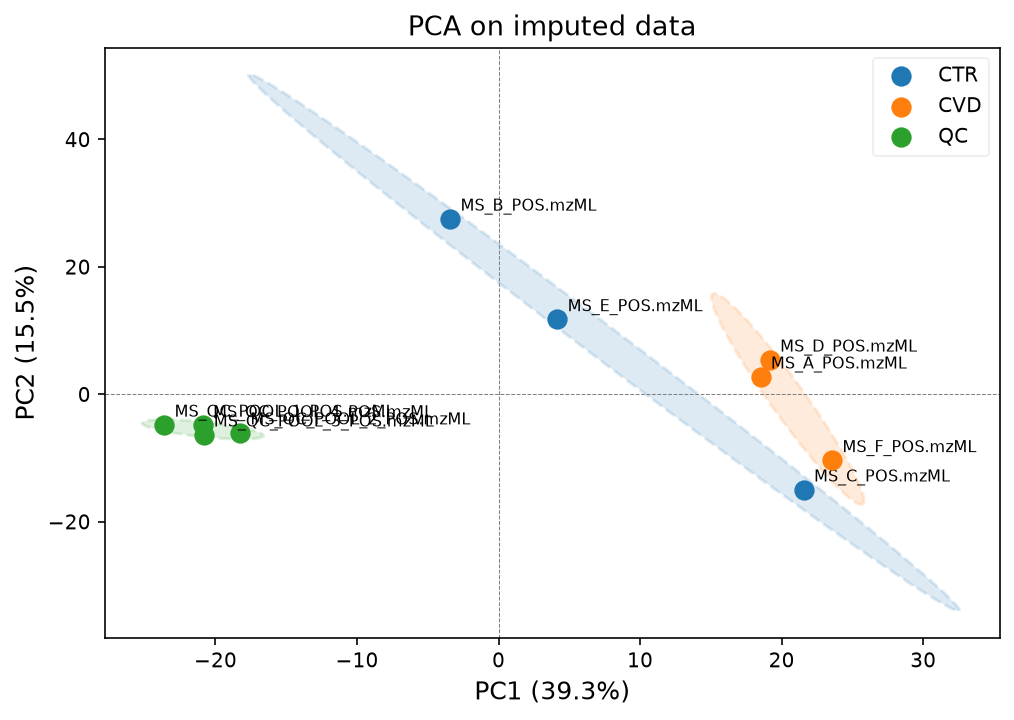
The first plot shows that the QC samples separate quite well from the biological samples.
The second plot shows that the two groups are also separated quite well, although one of the control samples, sample C, clusters slightly away from the rest.
Drift correction#
Now that we have a data frame that has been filtered and is fully filled in, we will look at the within-batch effects. In metabolomics, instrumental drift is common, so the signal degrades over time.
This is what we have the QC samples for, which is why they are so important to have included in the study.
We will test two different methods for correcting the instrumental drift in this notebook, inspect the results, and then choose one to continue with.
from acore import drift_correction as dc
Loess smoothing drift correction#
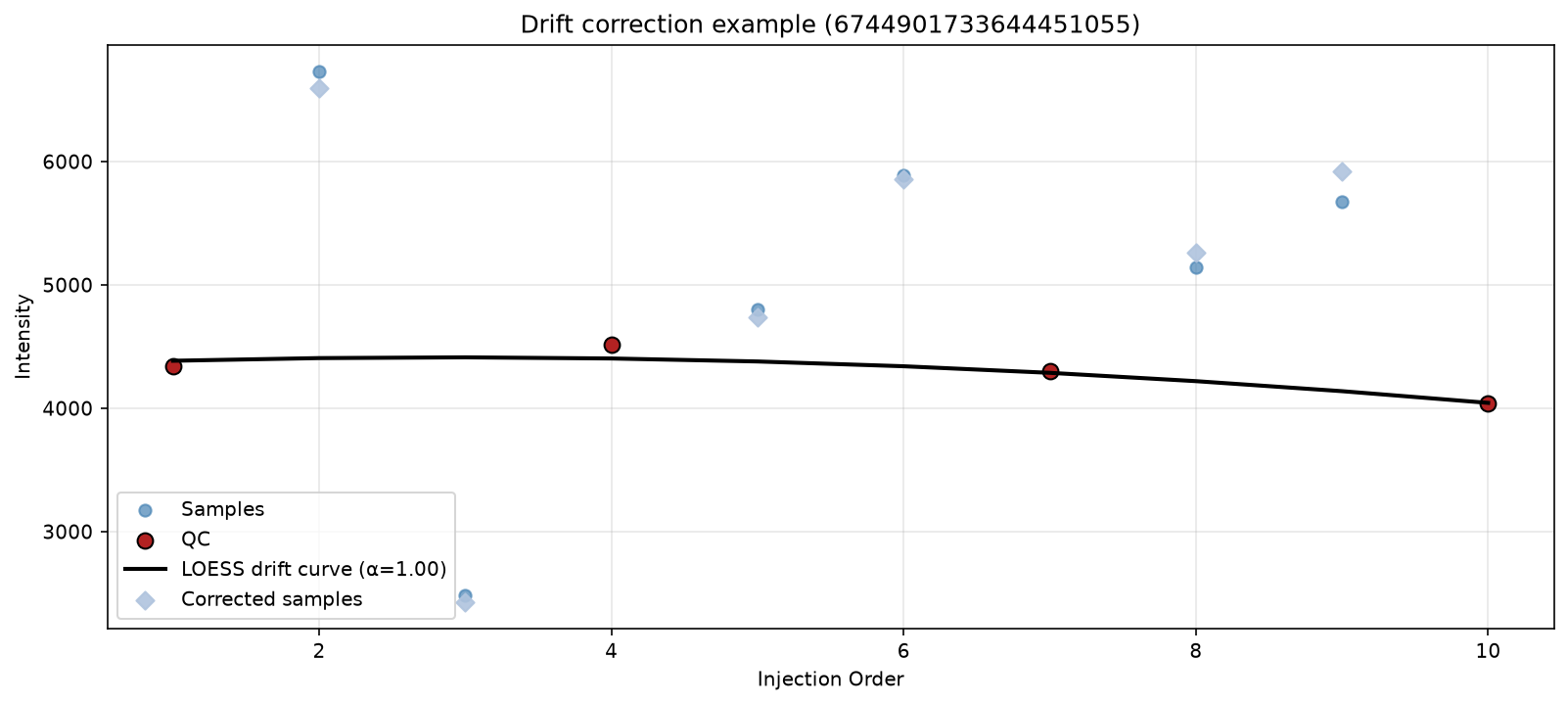
Pooled QC samples are injected at regular intervals over time throughout the experiment. The QC intensities for each features should theoretically be consistent in each measurement. Therefore, any variation in these samples reflect artificial variation, so instrumental drift, instead of biological variation.
LOESS (LOcally Estimated Scatterplot Smoothing) is a non-parametric regression that fits a smooth curve through data without assuming a fixed equation. We use it here to fit a curve over the QC points, and then we rescale the biological sample intensities by the drift estimate. This will be explained better by the visualisations in the real example below.
LOESS smoothing based drift correction is implemented in the loess_drift_correction acore module.
In order to use this method, we need to know the order in which the samples, including QCs, were run. We have this information in our metadata.
sample_order = pd.read_csv(fname_metadata).rename(
columns={"injection_index": "Sample ID", "derived_spectra_data_file": "File Name"}
)
sample_order
| File Name | phenotype | sample_name | age | Sample ID | |
|---|---|---|---|---|---|
| 0 | MS_QC_POOL_1_POS.mzML | QC | POOL1 | NaN | 1 |
| 1 | MS_A_POS.mzML | CVD | A | 53.000 | 2 |
| 2 | MS_B_POS.mzML | CTR | B | 30.000 | 3 |
| 3 | MS_QC_POOL_2_POS.mzML | QC | POOL2 | NaN | 4 |
| 4 | MS_C_POS.mzML | CTR | C | 66.000 | 5 |
| 5 | MS_D_POS.mzML | CVD | D | 36.000 | 6 |
| 6 | MS_QC_POOL_3_POS.mzML | QC | POOL3 | NaN | 7 |
| 7 | MS_E_POS.mzML | CTR | E | 66.000 | 8 |
| 8 | MS_F_POS.mzML | CVD | F | 44.000 | 9 |
| 9 | MS_QC_POOL_4_POS.mzML | QC | POOL4 | NaN | 10 |
data_corrected_loess, correction_info = dc.run_loess_drift_correction(
data=data_imputed, qc_rows=qcs, sample_rows=samples, sample_order=sample_order
)
data_corrected_loess
| id | 15167360593113340842 | 6744901733644451055 | 11872048905895506999 | 17709464339328039509 | 13922238524316478464 | 7261731678087078534 | 5009880893163685891 | 14568390051812142800 | 12965064019618390832 | 12018024353057911182 | ... | 10081236272997946842 | 11566491653082879729 | 1138090438525564760 | 16421084926908170456 | 15756805090958850402 | 6914550508003274460 | 8218613456253687867 | 1747965644941981794 | 8361543175644692425 | 9120899724421101875 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MS_D_POS.mzML | 143,573.432 | 5,855.581 | 11,405.280 | 20,793.801 | 11,825.461 | 1,844.135 | 12,982.441 | 65,407.523 | 45,379.148 | 6,168.096 | ... | 3,311.152 | 365.912 | 2,681.580 | 2,471.217 | 1,527.176 | 916.919 | 5,674.860 | 18,755.105 | 1,858.585 | 3,081.469 |
| MS_C_POS.mzML | 174,091.915 | 4,734.838 | 10,462.922 | 18,931.842 | 15,588.876 | 2,717.426 | 13,564.355 | 71,722.861 | 18,541.397 | 5,945.064 | ... | 9,868.868 | 933.982 | 1,654.844 | 837.204 | 1,790.423 | 1,434.580 | 4,974.297 | 17,793.996 | 1,676.537 | 1,407.971 |
| MS_E_POS.mzML | 89,312.250 | 5,260.916 | 10,546.469 | 21,125.835 | 17,346.741 | 2,590.945 | 12,254.776 | 52,116.191 | 56,595.381 | 5,561.124 | ... | 10,817.891 | 1,203.488 | 4,360.125 | 3,210.646 | 2,763.118 | 1,675.787 | 5,241.677 | 16,776.352 | 1,279.548 | 2,295.194 |
| MS_B_POS.mzML | 95,094.860 | 2,427.454 | 8,522.518 | 21,663.213 | 11,400.772 | 4,280.714 | 12,208.936 | 62,561.603 | 71,716.179 | 5,534.163 | ... | 13,814.247 | 940.708 | 4,004.152 | 2,886.022 | 2,259.722 | 1,182.188 | 3,222.823 | 16,529.271 | 6,600.248 | 1,684.365 |
| MS_A_POS.mzML | 161,510.647 | 6,595.786 | 9,987.575 | 19,345.144 | 19,090.120 | 2,322.102 | 13,335.245 | 67,622.219 | 55,492.351 | 5,954.525 | ... | 14,814.106 | 543.880 | 4,424.151 | 1,928.941 | 542.959 | 539.562 | 602.743 | 6,595.115 | 1,617.820 | 601.410 |
| MS_QC_POOL_1_POS.mzML | 204,976.266 | 4,269.719 | 9,339.026 | 23,969.714 | 10,213.482 | 3,403.048 | 11,393.676 | 56,809.333 | 32,456.642 | 5,281.738 | ... | 5,918.805 | 540.642 | 1,240.727 | 1,864.931 | 1,029.041 | 578.280 | 1,191.074 | 11,734.965 | 2,308.962 | 1,811.879 |
| MS_QC_POOL_4_POS.mzML | 205,505.990 | 4,311.723 | 9,363.215 | 23,794.615 | 11,036.823 | 3,487.608 | 11,311.188 | 58,189.891 | 32,756.643 | 5,272.373 | ... | 4,920.639 | 539.926 | 3,190.494 | 1,128.020 | 1,026.296 | 866.247 | 1,419.437 | 11,724.629 | 1,522.415 | 992.605 |
| MS_QC_POOL_3_POS.mzML | 205,634.870 | 4,328.721 | 9,445.987 | 24,190.450 | 10,084.861 | 3,510.502 | 11,570.338 | 56,498.883 | 31,830.424 | 5,352.694 | ... | 6,067.819 | 340.112 | 740.705 | 2,476.370 | 723.792 | 445.090 | 971.326 | 8,453.537 | 2,119.021 | 2,495.675 |
| MS_F_POS.mzML | 168,278.377 | 5,922.397 | 10,544.249 | 19,879.606 | 23,593.785 | 1,991.340 | 9,876.081 | 66,353.083 | 24,015.510 | 4,409.878 | ... | 7,393.029 | 497.741 | 662.707 | 2,076.658 | 1,197.659 | 1,085.413 | 7,661.980 | 14,042.548 | 1,211.548 | 1,986.662 |
| MS_QC_POOL_2_POS.mzML | 206,870.568 | 4,429.365 | 9,501.939 | 23,775.135 | 11,945.508 | 3,688.998 | 11,373.323 | 59,731.933 | 32,585.813 | 5,329.035 | ... | 4,050.263 | 335.325 | 4,790.817 | 799.673 | 708.475 | 1,099.972 | 1,427.385 | 8,398.979 | 999.685 | 669.995 |
10 rows × 897 columns
Our dataframe has the same structure as before.
We can also look at the object correction_info, if we want to trace the exact correction that was applied to every feature.
correction_info[data_corrected_loess.columns[0]]
{'alpha': np.float64(0.9999999999999999),
'drift_curve': [207528.80858189552,
207006.94258480993,
204507.5144035575,
200536.6918229344,
202029.39468897512,
205499.65973918274,
198870.40795967163,
203351.88080133556,
206331.68105175294,
207900.64328655135],
'y_qc': [198321.3, 204664.5, 206423.1, 207862.4],
'x_qc': [1, 4, 7, 10],
'rsd_qc': np.float64(1.7828199375522682),
'median': np.float64(205543.8),
'y_all': [169903.5,
89948.01,
173214.2,
157576.2,
93468.92,
143542.6,
198321.3,
204664.5,
206423.1,
207862.4],
'new_values': [168278.37668387496,
89312.2498549218,
174091.9153303282,
161510.64746873346,
95094.85997457383,
143573.43220580718,
204976.26590682287,
206870.56785178115,
205634.86986343015,
205505.99025435423],
'status': 'corrected'}
To understand better what is happening, we can plot a single feature with all its measurements over time, and the loess curve that is calculated over it. We can try out a few different features to see what would happen, and also try different smoothing parameters to see how the curve changes.
plot_loess_example_curve(
df=data_imputed,
feature_idx=1,
samples=samples,
qcs=qcs,
sample_order=sample_order,
)
CPCA#
Standard PCA finds orthogonal directions (principal components) that capture maximum variance in a single dataset. Common PCA extends this to multiple groups: instead of computing separate components per batch, it tries to find a set of components that are common across groups.
In this case, it finds the principal components that are common across all samples, because the assumption is that those are the ones that are due to artificial variation, whereas the variance in biological samples will not be shared across all.
First, we can plot a PCA for this purpose.
pca_for_cpca_drift(
data_imputed,
samples=samples, # list of col names, OR dict {group_name: [col names]}
qcs=qcs,
log_transform=True,
title="PCA",
)
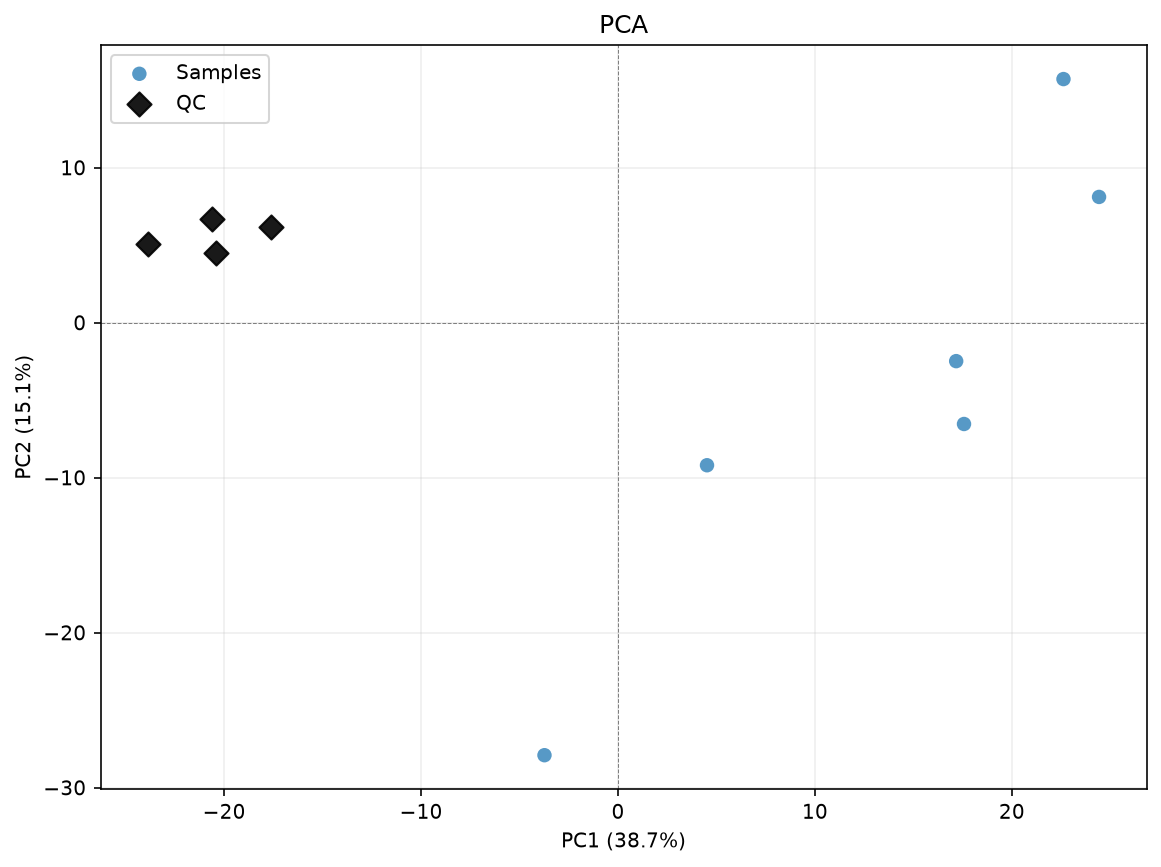
The QC samples are already quite close together, but there is some variation.
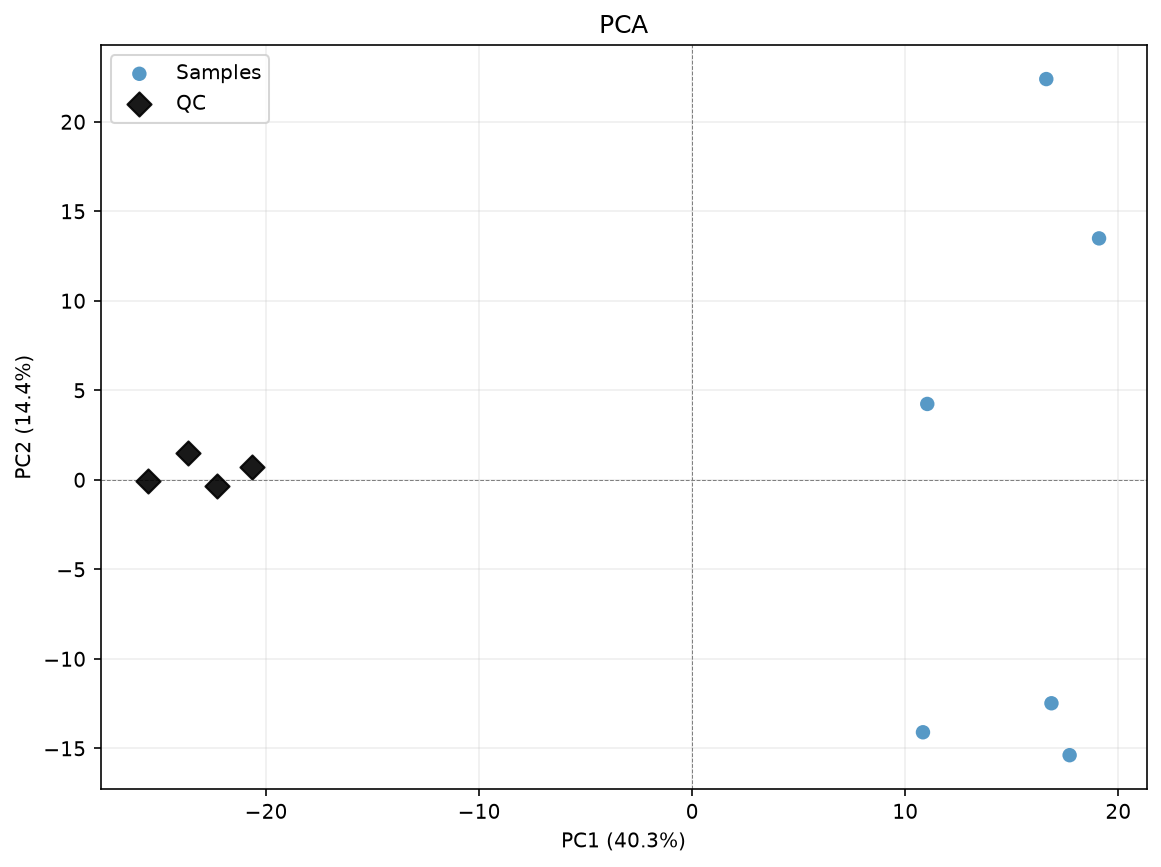
We can use the acore function cpca_drift_correction for this.
data_corrected_cpca = dc.run_cpca_drift_correction(
data_imputed, sample_rows=samples, qc_rows=qcs, n_comps=1
)
pca_for_cpca_drift(
data_corrected_cpca,
samples=samples, # list of col names, OR dict {group_name: [col names]}
qcs=qcs,
log_transform=True,
title="PCA",
)
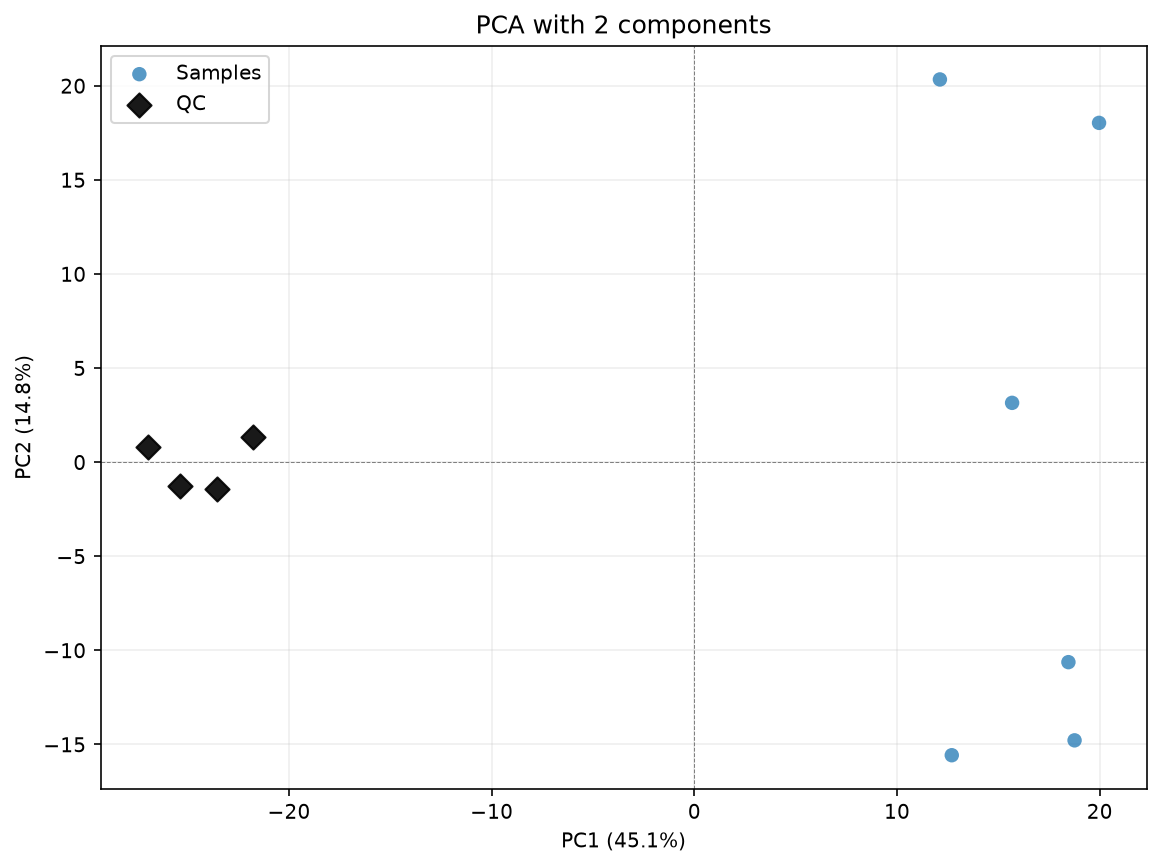
df_corrected_2comps = dc.run_cpca_drift_correction(
data_imputed, samples, qcs, n_comps=2
)
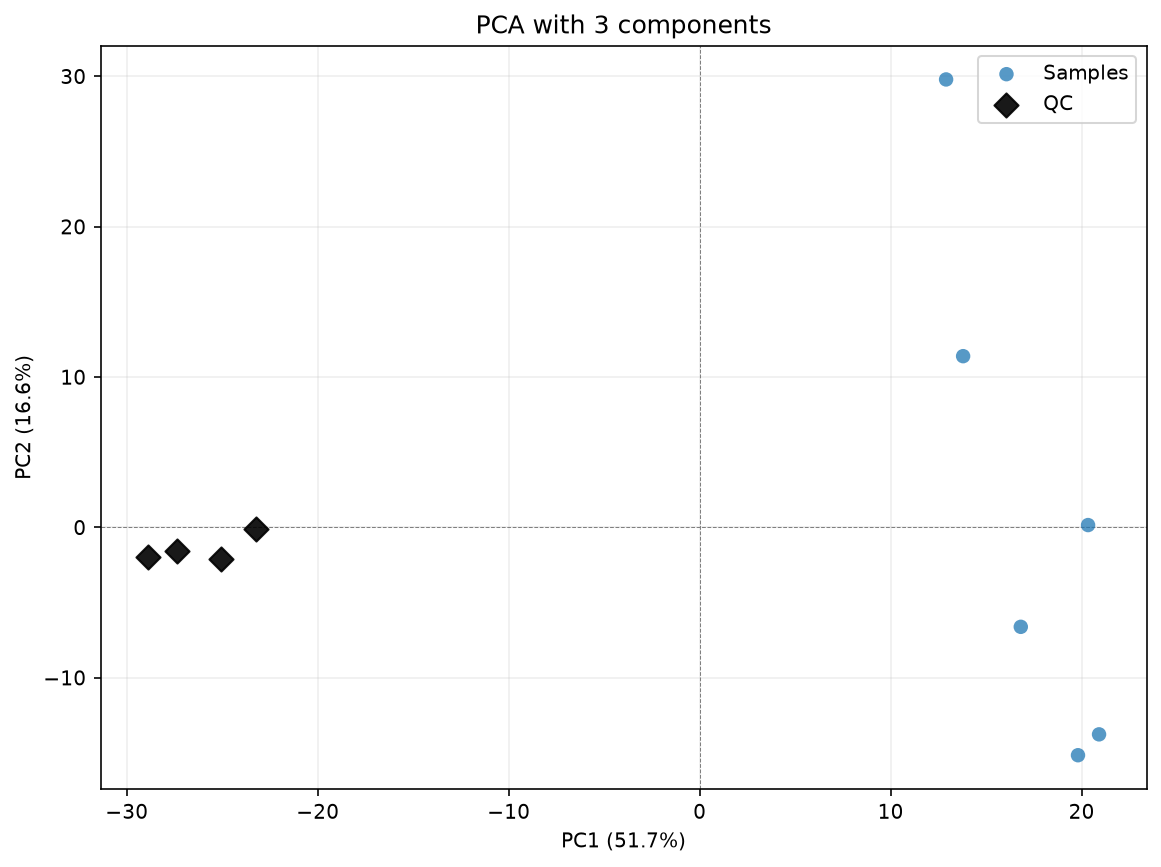
df_corrected_3comps = dc.run_cpca_drift_correction(
data_imputed, samples, qcs, n_comps=3
)
df_corrected_4comps = dc.run_cpca_drift_correction(
data_imputed, samples, qcs, n_comps=4
)
pca_for_cpca_drift(
df_corrected_2comps,
samples,
qcs,
log_transform=True,
title="PCA with 2 components",
)
pca_for_cpca_drift(
df_corrected_3comps,
samples,
qcs,
log_transform=True,
title="PCA with 3 components",
)
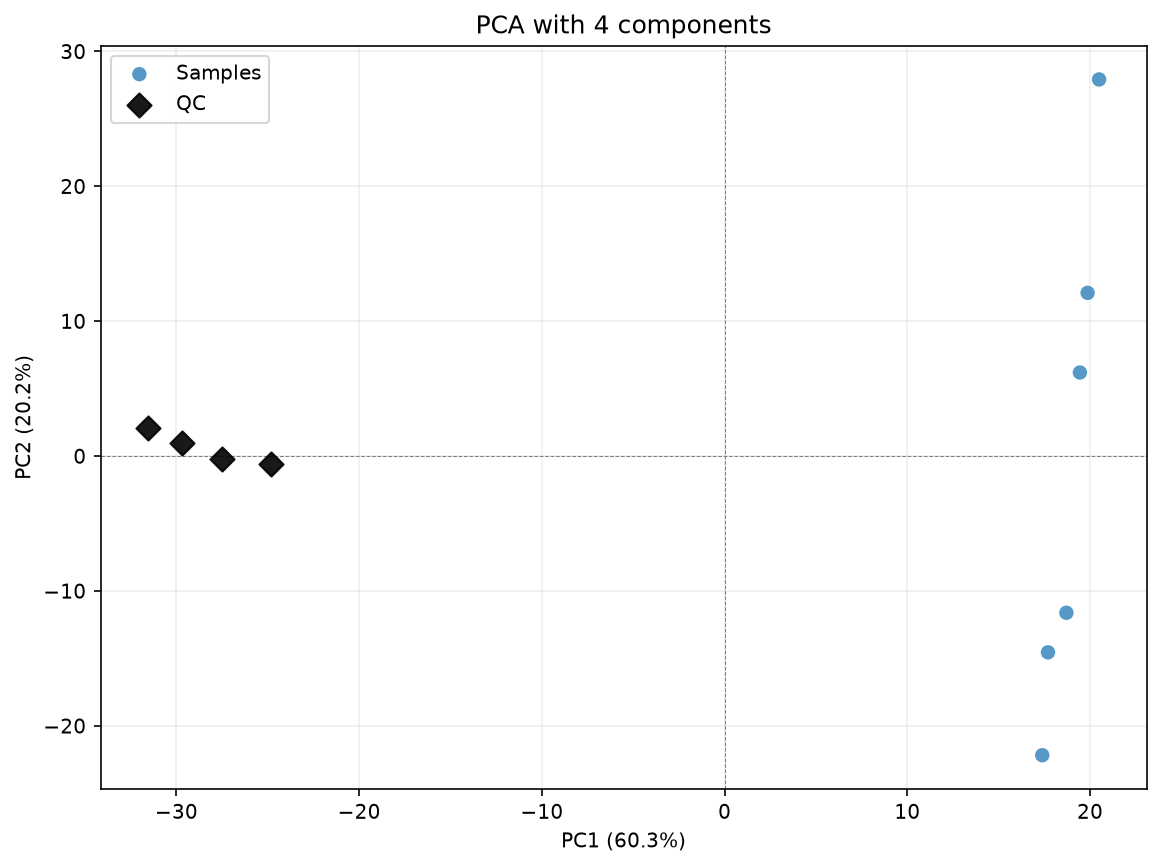
pca_for_cpca_drift(
df_corrected_4comps,
samples,
qcs,
log_transform=True,
title="PCA with 4 components",
)
pca_model, scores, var_explained = plot_pca(
data_corrected_loess,
groups_all,
label_points=True,
title="PCA on LOESS-corrected data",
)
pca_model, scores, var_explained = plot_pca(
data_corrected_loess,
groups_samples,
label_points=True,
title="PCA on LOESS-corrected data",
)
pca_model, scores, var_explained = plot_pca(
data_corrected_cpca,
groups_all,
label_points=True,
title="PCA on CPCA-corrected data",
)
pca_model, scores, var_explained = plot_pca(
data_corrected_cpca,
groups_samples,
label_points=True,
title="PCA on CPCA-corrected data",
)
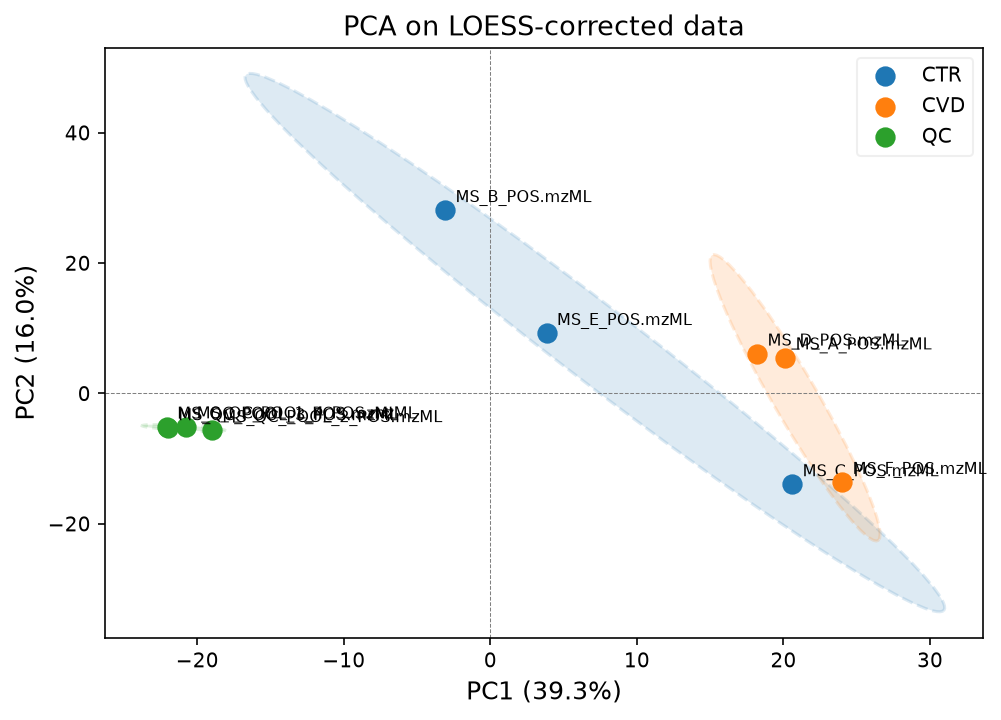
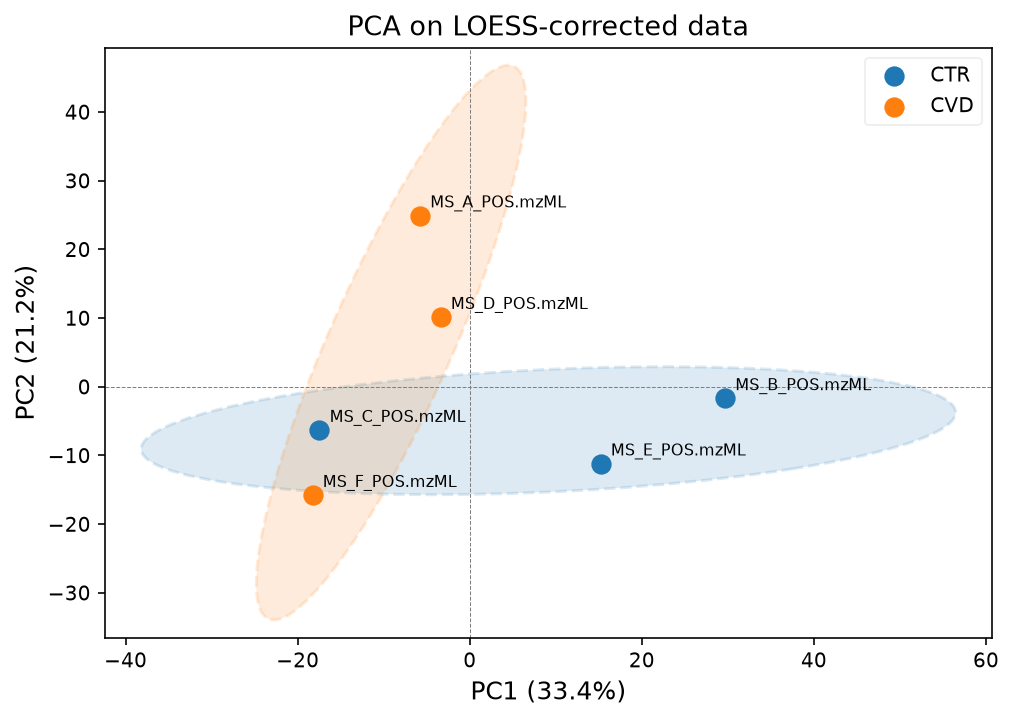
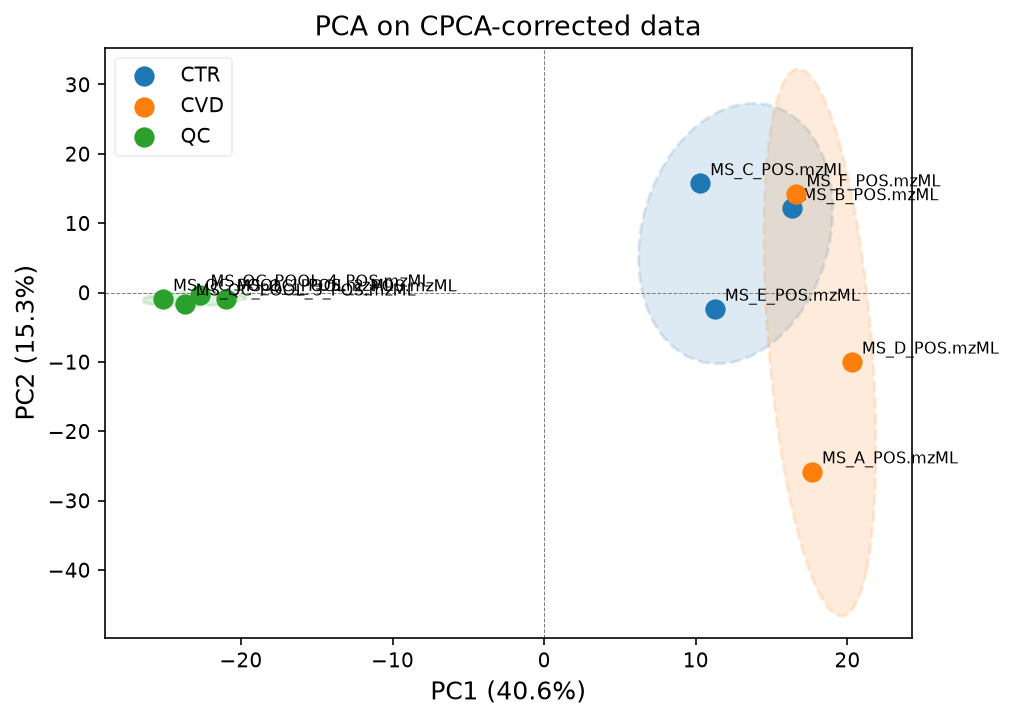
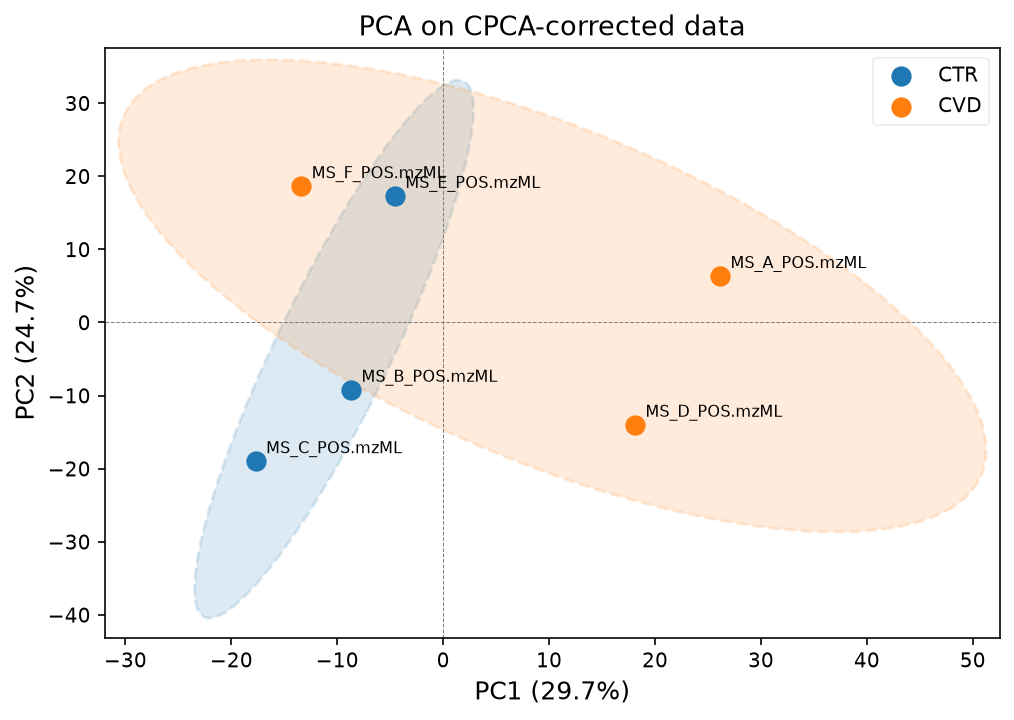
We will continue with the LOESS corrected data.
Normalization#
We will also normalise the data with Z-score normalisation. In this case, this will be mainly for visualisation purposes, again to see whether the data is separating well.
We will use an acore function for this again.
from acore import normalization
data_normalized = normalization.normalize_data(data_corrected_loess, "zscore")
data_normalized
| id | 15167360593113340842 | 6744901733644451055 | 11872048905895506999 | 17709464339328039509 | 13922238524316478464 | 7261731678087078534 | 5009880893163685891 | 14568390051812142800 | 12965064019618390832 | 12018024353057911182 | ... | 10081236272997946842 | 11566491653082879729 | 1138090438525564760 | 16421084926908170456 | 15756805090958850402 | 6914550508003274460 | 8218613456253687867 | 1747965644941981794 | 8361543175644692425 | 9120899724421101875 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MS_D_POS.mzML | 3.599 | -0.255 | -0.100 | 0.163 | -0.088 | -0.367 | -0.056 | 1.411 | 0.851 | -0.246 | ... | -0.326 | -0.409 | -0.344 | -0.350 | -0.376 | -0.393 | -0.260 | 0.106 | -0.367 | -0.333 |
| MS_C_POS.mzML | 4.191 | -0.261 | -0.111 | 0.112 | 0.024 | -0.314 | -0.029 | 1.500 | 0.102 | -0.230 | ... | -0.126 | -0.361 | -0.342 | -0.364 | -0.339 | -0.348 | -0.255 | 0.082 | -0.342 | -0.349 |
| MS_E_POS.mzML | 2.115 | -0.268 | -0.119 | 0.181 | 0.074 | -0.344 | -0.070 | 1.060 | 1.187 | -0.260 | ... | -0.111 | -0.383 | -0.294 | -0.327 | -0.339 | -0.370 | -0.269 | 0.058 | -0.381 | -0.353 |
| MS_B_POS.mzML | 2.247 | -0.325 | -0.156 | 0.209 | -0.076 | -0.274 | -0.054 | 1.344 | 1.598 | -0.239 | ... | -0.009 | -0.366 | -0.281 | -0.312 | -0.330 | -0.360 | -0.303 | 0.066 | -0.209 | -0.346 |
| MS_A_POS.mzML | 3.913 | -0.244 | -0.153 | 0.098 | 0.091 | -0.359 | -0.063 | 1.393 | 1.068 | -0.261 | ... | -0.024 | -0.407 | -0.303 | -0.370 | -0.407 | -0.407 | -0.405 | -0.244 | -0.378 | -0.405 |
| MS_QC_POOL_1_POS.mzML | 5.824 | -0.260 | -0.106 | 0.337 | -0.080 | -0.286 | -0.044 | 1.333 | 0.594 | -0.229 | ... | -0.210 | -0.373 | -0.352 | -0.333 | -0.358 | -0.372 | -0.353 | -0.034 | -0.320 | -0.335 |
| MS_QC_POOL_4_POS.mzML | 5.779 | -0.261 | -0.109 | 0.324 | -0.059 | -0.285 | -0.051 | 1.357 | 0.593 | -0.232 | ... | -0.242 | -0.374 | -0.294 | -0.356 | -0.359 | -0.364 | -0.348 | -0.038 | -0.344 | -0.360 |
| MS_QC_POOL_3_POS.mzML | 5.791 | -0.255 | -0.102 | 0.341 | -0.082 | -0.280 | -0.038 | 1.312 | 0.571 | -0.225 | ... | -0.203 | -0.375 | -0.363 | -0.311 | -0.364 | -0.372 | -0.356 | -0.131 | -0.322 | -0.310 |
| MS_F_POS.mzML | 4.348 | -0.243 | -0.112 | 0.152 | 0.257 | -0.354 | -0.131 | 1.466 | 0.269 | -0.286 | ... | -0.201 | -0.396 | -0.392 | -0.352 | -0.376 | -0.380 | -0.194 | -0.013 | -0.376 | -0.354 |
| MS_QC_POOL_2_POS.mzML | 5.698 | -0.256 | -0.107 | 0.313 | -0.035 | -0.278 | -0.052 | 1.370 | 0.572 | -0.230 | ... | -0.268 | -0.377 | -0.246 | -0.363 | -0.366 | -0.354 | -0.345 | -0.140 | -0.357 | -0.367 |
10 rows × 897 columns
plot_intensity_distribution(data_normalized)
pca_model, scores, var_explained = plot_pca(
data_normalized, groups_all, label_points=True, title="PCA on normalized data"
)
pca_model, scores, var_explained = plot_pca(
data_normalized, groups_samples, label_points=True, title="PCA on normalized data"
)
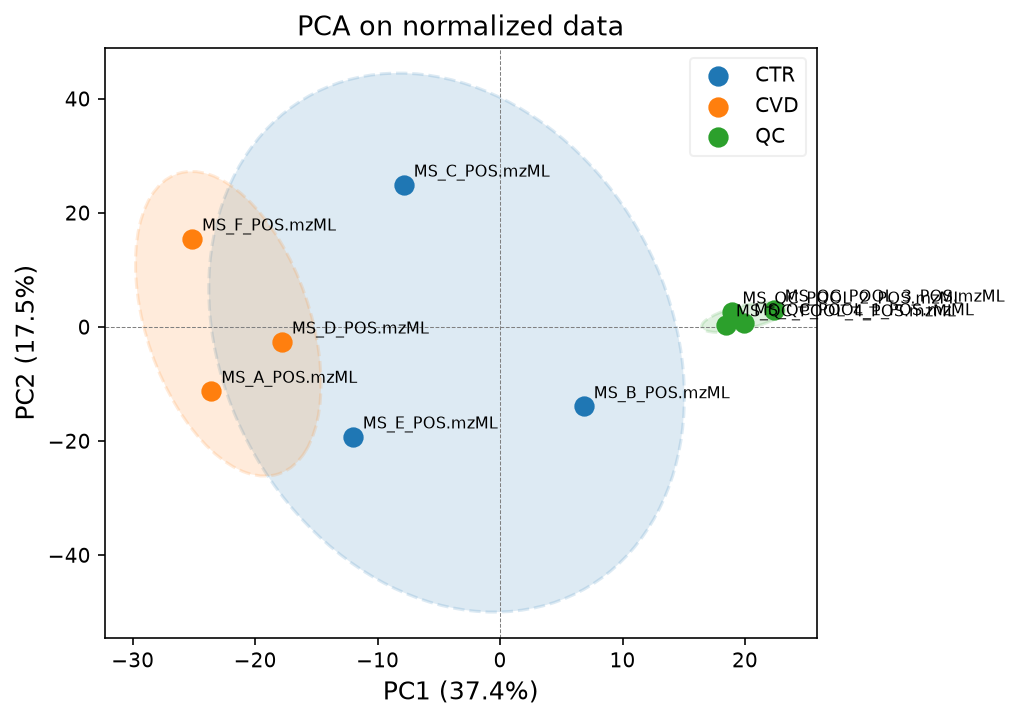
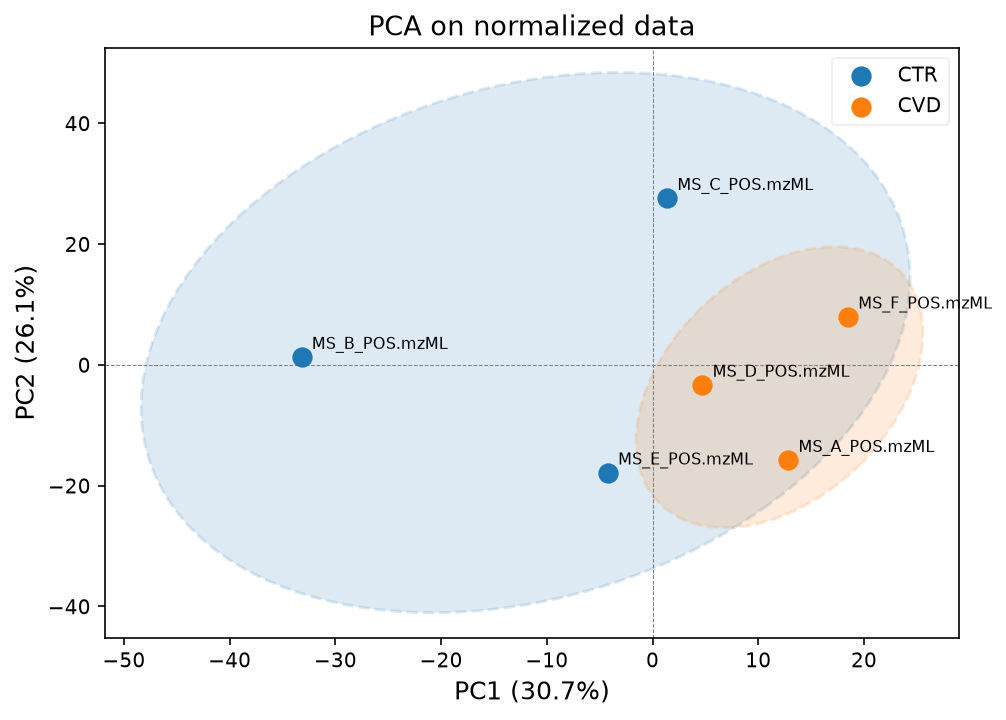
We can see that sample C is still distorting the separation. To know what went wrong with this control, we would need to know more about the sample preparation and background about the patients.
Statistical analysis#
ANCOVA#
We will now do a statistical analysis. We want to find out which ones of our metabolites are significantly more abundant in the cardiovascular disease group vs the control, or the other way around.
For this, we will do an ANCOVA (analysis of covariance), which compares group means on an outcome while statistically controlling for one or more continuous variables (covariates) that also influence the outcome. In this case, the covariate is age, because we have this in our metadata and know that it could potentially affect the sample outcomes.
We are using another acore function for this.
import acore.differential_regulation as ad
We need to do some preparation before we can run the function.
# Create the variable with the data
data_ancova = np.log2(data_imputed)
# Prepare the data to fit the function input
subject_col = data_ancova.index.name or "index"
data_ancova.rename_axis(subject_col, axis=0, inplace=True)
# Add group information to the data frame as a column
group_map = {
sample: label for label, samples in groups_all.items() for sample in samples
}
data_ancova.insert(0, "group", data_ancova.index.map(group_map))
# Remove the QC samples
data_ancova = data_ancova[data_ancova["group"] != "QC"]
# Add the column of age information
metadata = sample_order.copy().set_index("File Name")
data_ancova.insert(1, "age", metadata.loc[data_ancova.index, "age"])
data_ancova
| id | group | age | 15167360593113340842 | 6744901733644451055 | 11872048905895506999 | 17709464339328039509 | 13922238524316478464 | 7261731678087078534 | 5009880893163685891 | 14568390051812142800 | ... | 10081236272997946842 | 11566491653082879729 | 1138090438525564760 | 16421084926908170456 | 15756805090958850402 | 6914550508003274460 | 8218613456253687867 | 1747965644941981794 | 8361543175644692425 | 9120899724421101875 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| index | |||||||||||||||||||||
| MS_D_POS.mzML | CVD | 36.000 | 17.131 | 12.523 | 13.486 | 14.344 | 13.591 | 10.847 | 13.676 | 15.996 | ... | 11.762 | 8.101 | 11.481 | 11.242 | 10.256 | 9.895 | 12.408 | 13.888 | 11.005 | 11.554 |
| MS_C_POS.mzML | CTR | 66.000 | 17.402 | 12.229 | 13.360 | 14.211 | 13.987 | 11.385 | 13.730 | 16.121 | ... | 13.199 | 9.461 | 10.702 | 9.746 | 10.496 | 10.496 | 12.366 | 13.815 | 10.643 | 10.496 |
| MS_E_POS.mzML | CTR | 66.000 | 16.457 | 12.328 | 13.368 | 14.360 | 14.125 | 11.350 | 13.602 | 15.679 | ... | 13.711 | 10.121 | 12.337 | 11.470 | 11.341 | 10.852 | 11.963 | 13.949 | 10.805 | 10.960 |
| MS_B_POS.mzML | CTR | 30.000 | 16.512 | 11.277 | 13.051 | 14.406 | 13.505 | 11.967 | 13.554 | 15.900 | ... | 13.366 | 9.795 | 11.762 | 11.668 | 11.097 | 10.104 | 12.004 | 13.942 | 12.058 | 10.901 |
| MS_A_POS.mzML | CVD | 53.000 | 17.266 | 12.717 | 13.268 | 14.243 | 14.218 | 11.030 | 13.665 | 15.995 | ... | 13.284 | 9.264 | 11.751 | 11.163 | 9.256 | 8.895 | 9.702 | 12.815 | 9.643 | 9.496 |
| MS_F_POS.mzML | CVD | 44.000 | 17.374 | 12.471 | 13.361 | 14.267 | 14.549 | 10.963 | 13.291 | 16.029 | ... | 13.265 | 9.101 | 9.702 | 10.746 | 10.341 | 10.273 | 12.328 | 13.888 | 10.861 | 10.640 |
6 rows × 899 columns
Now we can run ANCOVA.
ancova = (
ad.run_ancova(
data_ancova.astype({"group": str}), # ! target needs to be of type str
# subject=subject_col, # not used
drop_cols=[],
group="group", # needs to be a string
covariates=["age"],
)
.set_index("identifier")
.sort_values(by="padj")
) # need to be floats?
ancova
| group1 | group2 | mean(group1) | std(group1) | mean(group2) | std(group2) | posthoc T-Statistics | posthoc pvalue | coef | std err | ... | log2FC | FC | F-statistics | pvalue | padj | correction | rejected | -log10 pvalue | Method | posthoc padj | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| identifier | |||||||||||||||||||||
| 122927701965791210 | CTR | CVD | 13.368 | 0.809 | 14.097 | 0.306 | 14.470 | 0.001 | 0.957 | 0.066 | ... | -0.729 | 0.603 | 209.372 | 0.001 | 0.194 | FDR correction BH | False | 3.145 | One-way ancova | 0.194 |
| 14183644523572007298 | CTR | CVD | 11.362 | 0.828 | 12.072 | 0.356 | 21.223 | 0.000 | 0.948 | 0.045 | ... | -0.710 | 0.611 | 450.404 | 0.000 | 0.194 | FDR correction BH | False | 3.640 | One-way ancova | 0.194 |
| 8688552057745683191 | CTR | CVD | 12.353 | 0.637 | 13.042 | 0.299 | 15.878 | 0.001 | 0.874 | 0.055 | ... | -0.689 | 0.620 | 252.110 | 0.001 | 0.194 | FDR correction BH | False | 3.265 | One-way ancova | 0.194 |
| 4803564648587047687 | CTR | CVD | 12.092 | 0.044 | 11.818 | 0.021 | -13.572 | 0.001 | -0.284 | 0.021 | ... | 0.274 | 1.209 | 184.207 | 0.001 | 0.194 | FDR correction BH | False | 3.063 | One-way ancova | 0.194 |
| 10207534598488119731 | CTR | CVD | 11.533 | 0.764 | 12.547 | 0.149 | 9.193 | 0.003 | 1.213 | 0.132 | ... | -1.013 | 0.495 | 84.513 | 0.003 | 0.318 | FDR correction BH | False | 2.565 | One-way ancova | 0.318 |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| 7068176369469578268 | CTR | CVD | 11.981 | 0.083 | 11.974 | 0.242 | -0.023 | 0.983 | -0.004 | 0.175 | ... | 0.007 | 1.005 | 0.001 | 0.983 | 0.992 | FDR correction BH | False | 0.007 | One-way ancova | 0.992 |
| 18162318233842994935 | CTR | CVD | 13.669 | 0.374 | 13.720 | 0.325 | -0.012 | 0.991 | -0.004 | 0.308 | ... | -0.051 | 0.965 | 0.000 | 0.991 | 0.994 | FDR correction BH | False | 0.004 | One-way ancova | 0.994 |
| 3927519029408433709 | CTR | CVD | 13.941 | 0.112 | 13.919 | 0.142 | -0.009 | 0.993 | -0.001 | 0.111 | ... | 0.022 | 1.015 | 0.000 | 0.993 | 0.994 | FDR correction BH | False | 0.003 | One-way ancova | 0.994 |
| 13146520139614759536 | CTR | CVD | 11.517 | 0.213 | 11.555 | 0.121 | 0.010 | 0.993 | 0.001 | 0.138 | ... | -0.038 | 0.974 | 0.000 | 0.993 | 0.994 | FDR correction BH | False | 0.003 | One-way ancova | 0.994 |
| 491936035475549414 | CTR | CVD | 11.796 | 2.677 | 12.144 | 2.116 | 0.005 | 0.996 | 0.010 | 2.165 | ... | -0.348 | 0.786 | 0.000 | 0.996 | 0.996 | FDR correction BH | False | 0.002 | One-way ancova | 0.996 |
897 rows × 22 columns
We have filtered the table by the adjusted pvalue. We can look at the top values to see how good our best hits are. We are sorting the data by the adjusted pvalue to see what our best hits are, and how good they actually are. In this case, our best hits have adjusted pvalues of 0.321 and higher, which is quite high, and not significant.
We can inspect the results in a few different ways. First of all, we can look at the group averages, which are shown in the first six columns.
ancova.iloc[:, :6]
| group1 | group2 | mean(group1) | std(group1) | mean(group2) | std(group2) | |
|---|---|---|---|---|---|---|
| identifier | ||||||
| 122927701965791210 | CTR | CVD | 13.368 | 0.809 | 14.097 | 0.306 |
| 14183644523572007298 | CTR | CVD | 11.362 | 0.828 | 12.072 | 0.356 |
| 8688552057745683191 | CTR | CVD | 12.353 | 0.637 | 13.042 | 0.299 |
| 4803564648587047687 | CTR | CVD | 12.092 | 0.044 | 11.818 | 0.021 |
| 10207534598488119731 | CTR | CVD | 11.533 | 0.764 | 12.547 | 0.149 |
| ... | ... | ... | ... | ... | ... | ... |
| 7068176369469578268 | CTR | CVD | 11.981 | 0.083 | 11.974 | 0.242 |
| 18162318233842994935 | CTR | CVD | 13.669 | 0.374 | 13.720 | 0.325 |
| 3927519029408433709 | CTR | CVD | 13.941 | 0.112 | 13.919 | 0.142 |
| 13146520139614759536 | CTR | CVD | 11.517 | 0.213 | 11.555 | 0.121 |
| 491936035475549414 | CTR | CVD | 11.796 | 2.677 | 12.144 | 2.116 |
897 rows × 6 columns
If we filter to the words below, we can inspect the test results (based on a linear model) for each feature (on each row). The posthoc values are not interesting in this case as we are only comparing between two groups (ctr and cvd). We are mainly interested in the adjusted pvalue (pajd), and in whether the null hypothesis was rejected or not. If rejected=True, it means our feature was significant according to the thresholds we have set.
regex_filter = "pval|padj|reject|post"
ancova.filter(regex=regex_filter)
| posthoc T-Statistics | posthoc pvalue | pvalue | padj | rejected | -log10 pvalue | posthoc padj | |
|---|---|---|---|---|---|---|---|
| identifier | |||||||
| 122927701965791210 | 14.470 | 0.001 | 0.001 | 0.194 | False | 3.145 | 0.194 |
| 14183644523572007298 | 21.223 | 0.000 | 0.000 | 0.194 | False | 3.640 | 0.194 |
| 8688552057745683191 | 15.878 | 0.001 | 0.001 | 0.194 | False | 3.265 | 0.194 |
| 4803564648587047687 | -13.572 | 0.001 | 0.001 | 0.194 | False | 3.063 | 0.194 |
| 10207534598488119731 | 9.193 | 0.003 | 0.003 | 0.318 | False | 2.565 | 0.318 |
| ... | ... | ... | ... | ... | ... | ... | ... |
| 7068176369469578268 | -0.023 | 0.983 | 0.983 | 0.992 | False | 0.007 | 0.992 |
| 18162318233842994935 | -0.012 | 0.991 | 0.991 | 0.994 | False | 0.004 | 0.994 |
| 3927519029408433709 | -0.009 | 0.993 | 0.993 | 0.994 | False | 0.003 | 0.994 |
| 13146520139614759536 | 0.010 | 0.993 | 0.993 | 0.994 | False | 0.003 | 0.994 |
| 491936035475549414 | 0.005 | 0.996 | 0.996 | 0.996 | False | 0.002 | 0.996 |
897 rows × 7 columns
As we could already see by the high adjusted pvalues in the data frame, none of the values are significant, which is why rejected=False for all of them.
You can also look at the rest of the results.
ancova.iloc[:, 6:].filter(regex=f"^(?!.*({regex_filter})).*$")
| coef | std err | Conf. Int. Low | Conf. Int. Upp. | log2FC | FC | F-statistics | correction | Method | |
|---|---|---|---|---|---|---|---|---|---|
| identifier | |||||||||
| 122927701965791210 | 0.957 | 0.066 | 0.746 | 1.167 | -0.729 | 0.603 | 209.372 | FDR correction BH | One-way ancova |
| 14183644523572007298 | 0.948 | 0.045 | 0.806 | 1.090 | -0.710 | 0.611 | 450.404 | FDR correction BH | One-way ancova |
| 8688552057745683191 | 0.874 | 0.055 | 0.699 | 1.049 | -0.689 | 0.620 | 252.110 | FDR correction BH | One-way ancova |
| 4803564648587047687 | -0.284 | 0.021 | -0.351 | -0.218 | 0.274 | 1.209 | 184.207 | FDR correction BH | One-way ancova |
| 10207534598488119731 | 1.213 | 0.132 | 0.793 | 1.633 | -1.013 | 0.495 | 84.513 | FDR correction BH | One-way ancova |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| 7068176369469578268 | -0.004 | 0.175 | -0.560 | 0.552 | 0.007 | 1.005 | 0.001 | FDR correction BH | One-way ancova |
| 18162318233842994935 | -0.004 | 0.308 | -0.984 | 0.977 | -0.051 | 0.965 | 0.000 | FDR correction BH | One-way ancova |
| 3927519029408433709 | -0.001 | 0.111 | -0.355 | 0.353 | 0.022 | 1.015 | 0.000 | FDR correction BH | One-way ancova |
| 13146520139614759536 | 0.001 | 0.138 | -0.438 | 0.441 | -0.038 | 0.974 | 0.000 | FDR correction BH | One-way ancova |
| 491936035475549414 | 0.010 | 2.165 | -6.881 | 6.901 | -0.348 | 0.786 | 0.000 | FDR correction BH | One-way ancova |
897 rows × 9 columns
Now we can plot a volcano plot, which is a scatter plot that combines statistical significance and effect size (magnitude of change) in a single view.
scatter_plot_adv = vuecore.plots.basic.scatter.create_scatter_plot(
data=ancova.reset_index(),
x="log2FC",
y="-log10 pvalue",
color="rejected",
title="Volcano Plot showing CTR vs CVD samples",
subtitle="Visualizing ANCOVA results",
labels={
"log2FC": "Log2 Fold Change",
"-log10 pvalue": "-log10(p-value)",
"pvalue": "Raw P value",
"rejected": "FDR corrected Significant",
"identifier": "Feature Identifier",
},
hover_data=["identifier"],
# currently does not work:
# color_discrete_map={False: "#2166AC", True: "#B2182B"}, # Blue # Red
color_discrete_sequence=["red", "blue"],
opacity=1,
marker_line_width=1,
marker_line_color="darkgray",
width=800,
height=600,
)
scatter_plot_adv
If we had significant values, they would show up in blue in this plot.
fig, axes = plt.subplots(1, 2, figsize=(12, 5))
# Raw p-values
axes[0].hist(
ancova["pvalue"].dropna(),
bins=50,
color="steelblue",
edgecolor="white",
linewidth=0.5,
)
axes[0].set_title("Distribution of Raw P-values", fontsize=14, fontweight="bold")
axes[0].set_xlabel("Raw p-value")
axes[0].set_ylabel("Count")
axes[0].axvline(x=0.05, color="red", linestyle="--", linewidth=1.5, label="α = 0.05")
axes[0].legend()
# Adjusted p-values
axes[1].hist(
ancova["padj"].dropna(),
bins=50,
color="darkorange",
edgecolor="white",
linewidth=0.5,
)
axes[1].set_title(
"Distribution of Adjusted P-values (FDR correction BH)",
fontsize=14,
fontweight="bold",
)
axes[1].set_xlabel("Adjusted p-value")
axes[1].set_ylabel("Count")
axes[1].axvline(x=0.05, color="red", linestyle="--", linewidth=1.5, label="α = 0.05")
axes[1].legend()
plt.suptitle("P-value Distributions", fontsize=16, fontweight="bold", y=1.02)
plt.tight_layout()
plt.show()
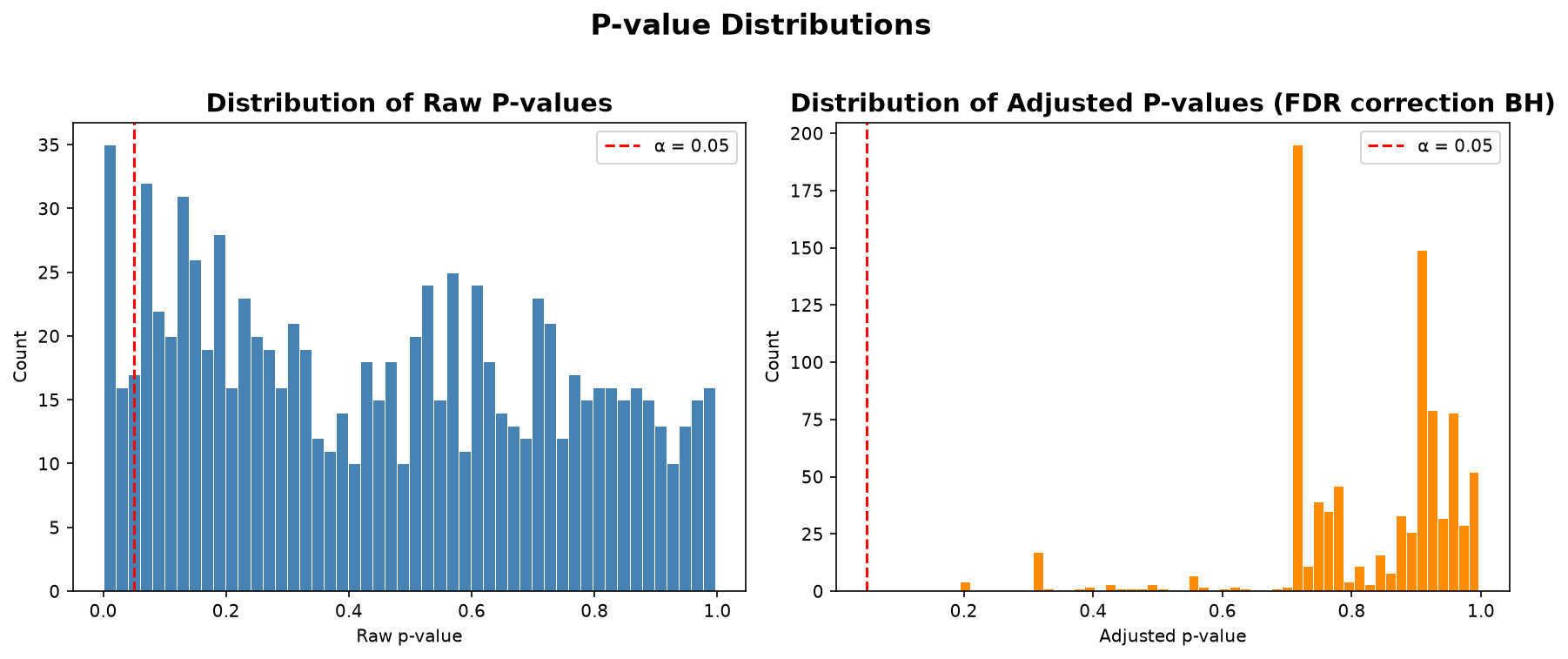
In the above plot, we are looking at our pvalues and adjusted pvalues in more detail.
We can also see how many features survived each threshold in the calculations below.
print("Raw p < 0.05:", (ancova["pvalue"] < 0.05).sum())
print("Raw p < 0.01:", (ancova["pvalue"] < 0.01).sum())
print("padj < 0.05: ", (ancova["padj"] < 0.05).sum())
print("padj < 0.1: ", (ancova["padj"] < 0.1).sum())
Raw p < 0.05: 58
Raw p < 0.01: 23
padj < 0.05: 0
padj < 0.1: 0
Identify hits#
Finally, we can identify our best hits to then be analysed further. Once we have found our top hits, we can match the (for now unknown) features with library spectra and identify which metabolites they are. Then, we can carry out further analyses such as enrichment analysis.
We can choose some thresholds for this, often we would choose an adjusted pvalue of 0.05 or lower, and an absolute log2 fold change of 1 or higher.
# Primary hit list for follow-up
hits = ancova[(ancova["padj"] < 0.05) & (ancova["log2FC"].abs() > 1)]
print(f"Candidate metabolites: {len(hits)}")
print(f"Upregulated in CVD: {(hits['log2FC'] > 0).sum()}")
print(f"Downregulated in CVD: {(hits['log2FC'] < 0).sum()}")
Candidate metabolites: 0
Upregulated in CVD: 0
Downregulated in CVD: 0
We can save and export our hits to a csv file. Usually we would save the top hits and explore them further, but in this case, we will save the whole ancova, and just look further into the top metabolites here, although they are not significant.
ancova.to_csv("results_prepared/ancova_results.csv")
Done!