6.4 Part 2 — Gene Set Enrichment Analysis (GSEA)
What is GSEA?
GSEA asks: “Are genes in a pathway coordinately shifting up or down across my full ranked list?”
Unlike ORA, GSEA:
- Uses all tested genes — capturing coordinated but subtle pathway-level changes
- Ranks genes by a metric encoding both significance and direction
- Returns a Normalised Enrichment Score (NES): positive NES = pathway upregulated in treatment; negative NES = pathway downregulated
Why signed log p-value for ranking?
rank = sign(LFC) x -log10(pvalue)
Genes at the top are significantly upregulated; genes at the bottom are significantly downregulated. This is more robust than ranking on LFC alone, which can place noisy high-fold-change genes from lowly expressed genes at the extremes.
📘 Note: Genes with pvalue = 0 (below machine precision in DESeq2) are excluded
because -log10(0) = Inf is not a valid ranking score. These are the most extremely
significant genes and their exclusion has minimal impact on pathway-level results.
6.4.1 Prepare the Ranked Gene List
ranked_df <- res_df_sym %>%
filter(!is.na(pvalue), !is.na(log2FoldChange)) %>%
filter(pvalue > 0) %>% # exclude p=0 -> log10(0) = Inf
mutate(rank_metric = sign(log2FoldChange) * -log10(pvalue)) %>%
arrange(desc(rank_metric)) %>%
distinct(gene, .keep_all = TRUE)
rank_vector <- ranked_df$rank_metric
names(rank_vector) <- ranked_df$gene
cat("Genes in ranked list :", length(rank_vector), "\n")## Genes in ranked list : 3697## Infinite values : 0## Top 5 (upregulated) : b3508, b3510, gadC, b3509, gadB## Bottom 5 (downreg.) : mglB, garL, b3127, b0553, garR6.4.2 Convert GMT to fgsea Format
fgsea requires a named list of gene vectors — one entry per pathway.
pathways_list <- setNames(
kegg_gmt$list_of_values,
kegg_gmt$ontology_name
)
cat("Pathways in list:", length(pathways_list), "\n")## Pathways in list: 121## Example entry : aceE aceF lpd yahK frmA6.4.3 Run GSEA
set.seed(42)
gsea_results <- fgsea(
pathways = pathways_list,
stats = rank_vector,
minSize = 10,
maxSize = 500,
nPermSimple = 10000
)
cat("Pathways tested :", nrow(gsea_results), "\n")## Pathways tested : 77## Significant (padj < 0.05): 2## Nominal (pval < 0.05) : 186.4.4 Filter and Inspect Significant Pathways
gsea_sig <- gsea_results %>%
as.data.frame() %>%
filter(padj < 0.05) %>%
arrange(padj)
cat("Significant GSEA pathways:", nrow(gsea_sig), "\n")## Significant GSEA pathways: 2## Activated (NES > 0) : 0## Suppressed (NES < 0) : 2DT::datatable(
gsea_sig %>%
select(pathway, NES, pval, padj, size) %>%
mutate(
direction = ifelse(NES > 0, "Activated", "Suppressed"),
across(where(is.numeric), \(x) round(x, 4))
) %>%
arrange(desc(abs(NES))),
rownames = FALSE,
colnames = c("Pathway", "NES", "p-value", "padj", "Size", "Direction"),
extensions = c("Buttons", "Scroller"),
options = list(
dom = "Bfrtip",
buttons = c("copy", "csv"),
scrollX = TRUE,
scrollY = 300,
scroller = TRUE
),
caption = "Significant GSEA pathways — treatment vs control (padj < 0.05)"
)Visualise GSEA Results
6.4.5 NES Bar Plot — Activated and Suppressed Pathways
cols_direction <- c("Activated" = "#d7191c", "Suppressed" = "#2c7bb6")
# Use top 20 by |NES| if more than 20 significant, otherwise show all
gsea_bar <- gsea_results %>%
as.data.frame() %>%
filter(pval < 0.05) %>%
slice_max(abs(NES), n = 20) %>%
mutate(
direction = ifelse(NES > 0, "Activated", "Suppressed"),
pathway = fct_reorder(pathway, NES)
)
gsea_nesplot <- ggplot(gsea_bar,
aes(x = NES,
y = pathway,
fill = direction)) +
geom_col(width = 0.7, alpha = 0.9) +
geom_vline(xintercept = 0, color = "black", linewidth = 0.5) +
geom_vline(xintercept = c(-1, 1), linetype = "dashed",
color = "grey50", linewidth = 0.4) +
scale_fill_manual(values = cols_direction) +
theme_bw() +
theme(legend.position = "bottom") +
labs(
title = "GSEA — Normalised Enrichment Scores (pval < 0.05)",
subtitle = "Treatment vs Control | E. coli MG1655",
x = "Normalised Enrichment Score (NES)",
y = NULL,
fill = NULL
)
gsea_nesplot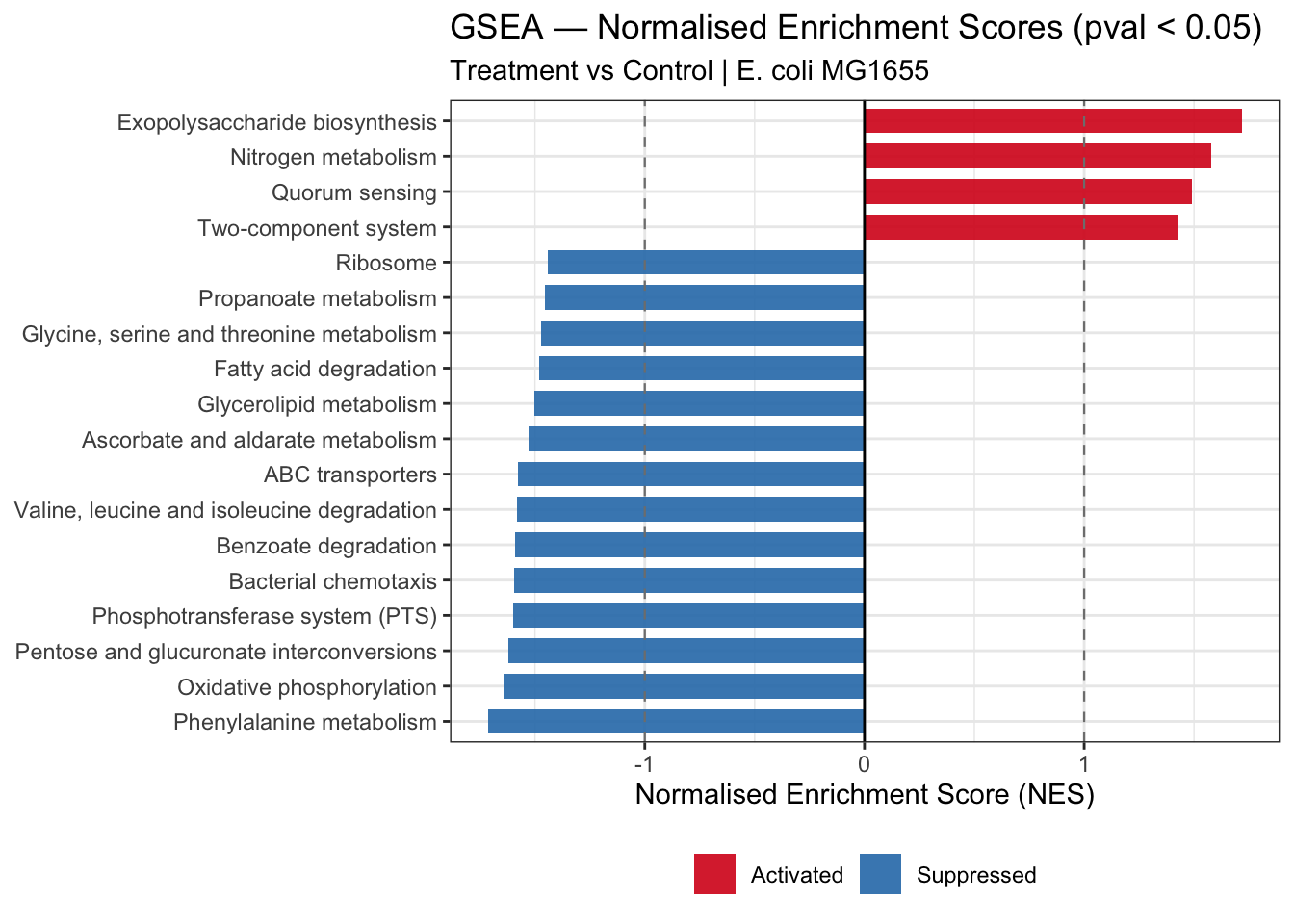
6.4.6 Dot Plot — NES, padj and Gene Set Size
gsea_dotplot <- ggplot(gsea_bar,
aes(x = NES,
y = pathway,
size = size,
color = padj)) +
geom_point(alpha = 0.85) +
geom_vline(xintercept = 0, linetype = "dashed", color = "grey50") +
scale_color_gradient(low = "#CA0020", high = "#92C5DE", name = "padj") +
scale_size_continuous(name = "Genes in set", range = c(3, 10)) +
theme_bw() +
theme(legend.position = "right") +
labs(
title = "GSEA — Pathway Dot Plot",
subtitle = "Point size = genes in set | Colour = padj",
x = "NES",
y = NULL
)
gsea_dotplot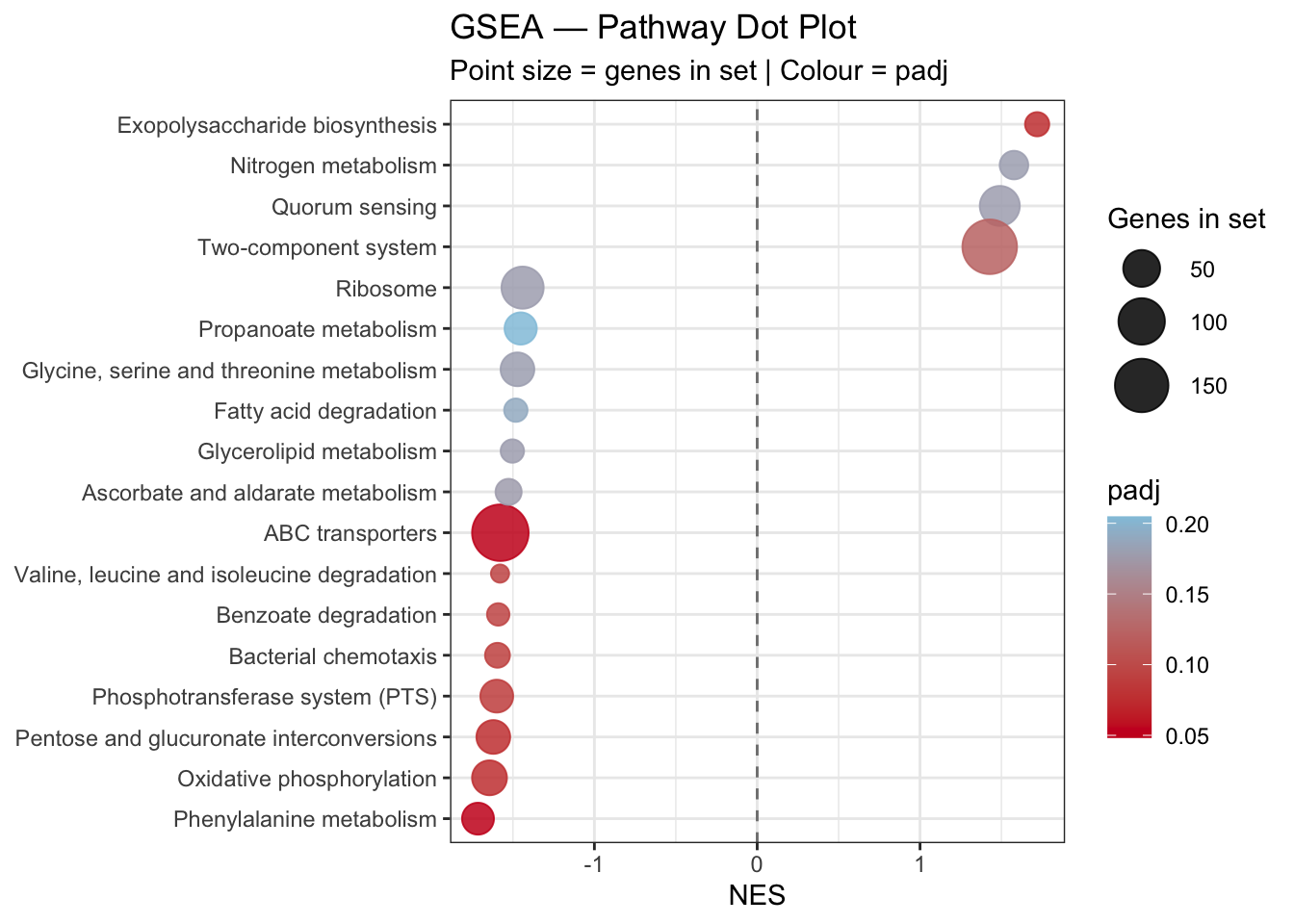
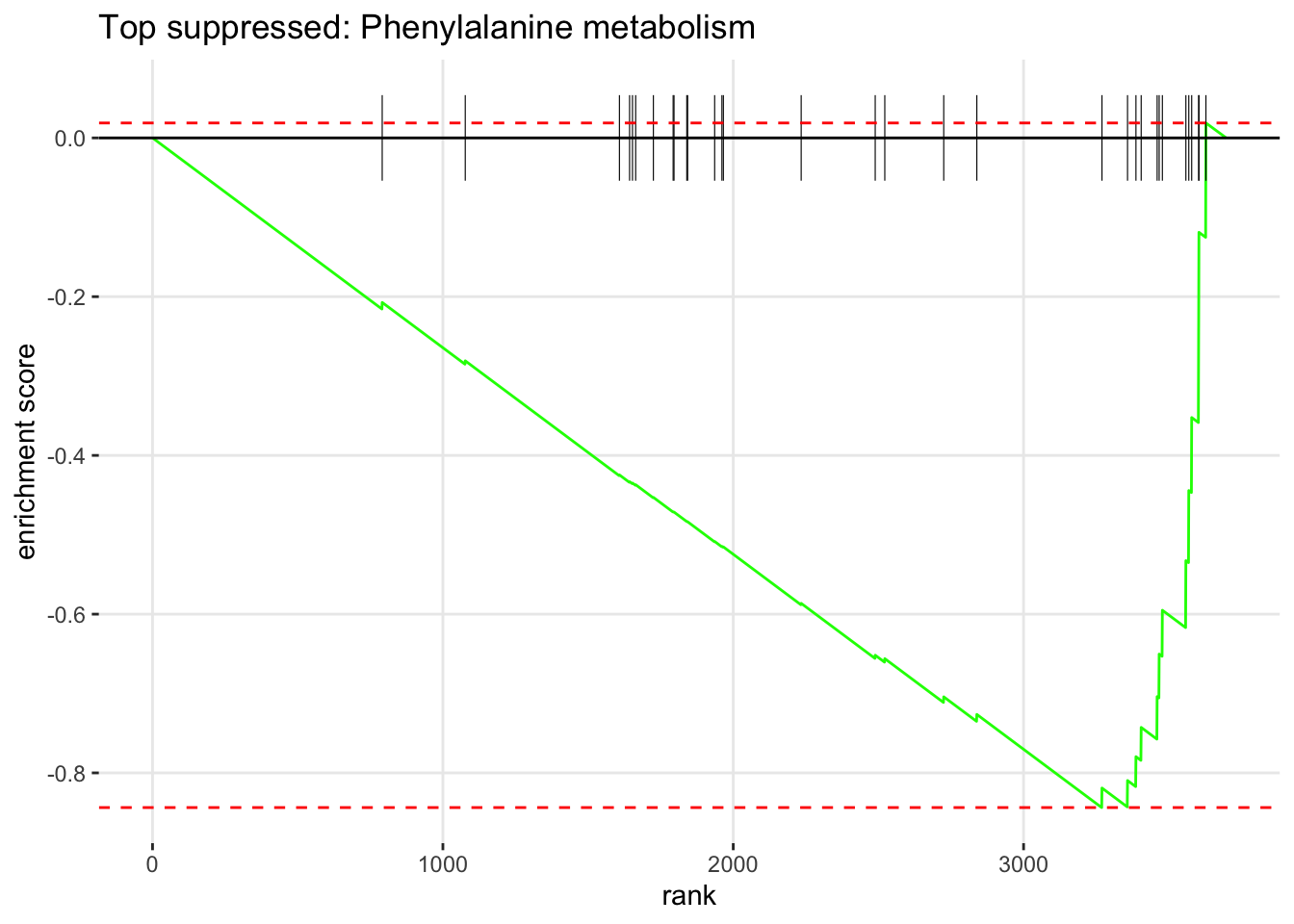
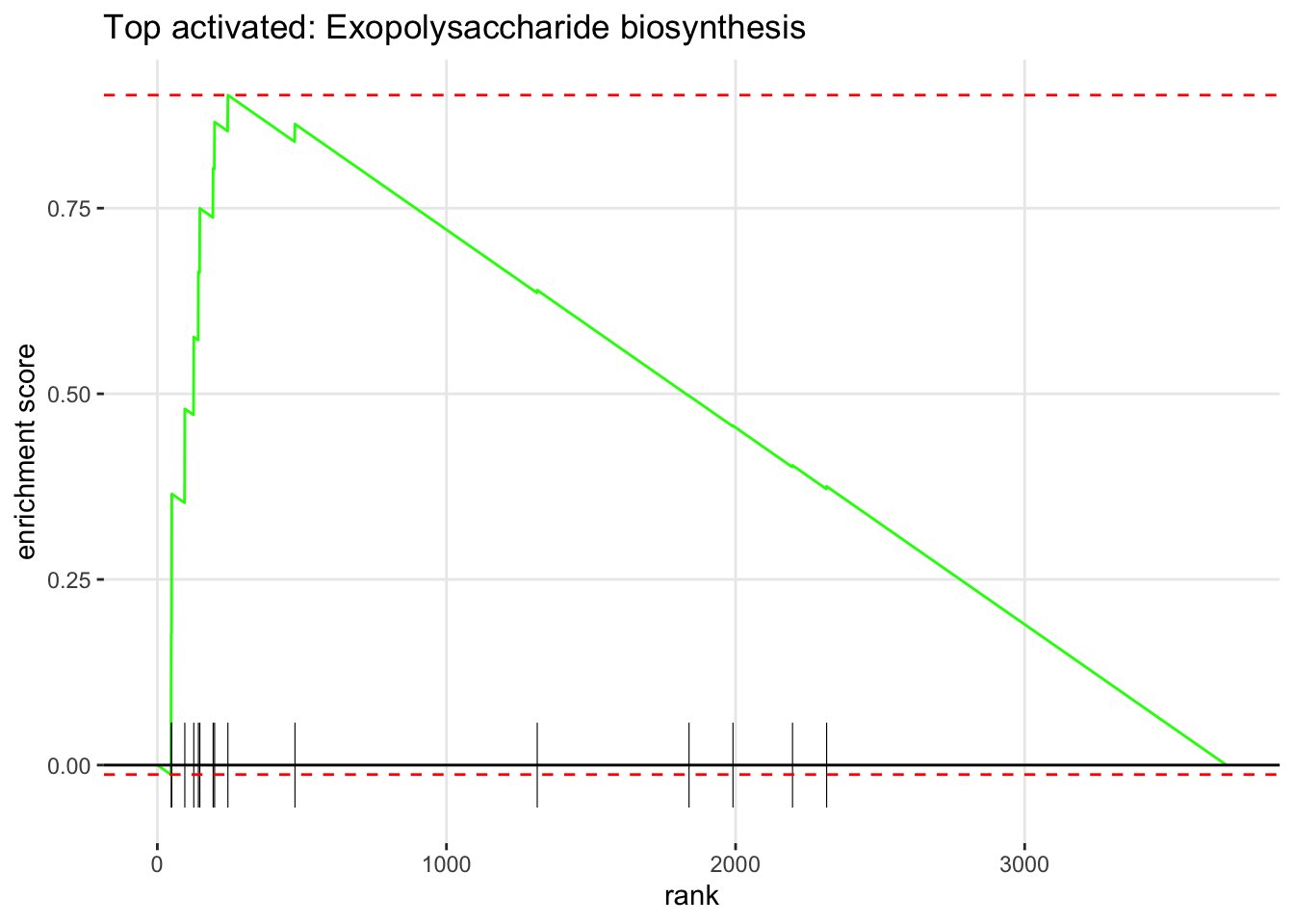
6.4.7 Enrichment Plot — Top Pathways
The enrichment plot shows the running sum of the enrichment score for a single pathway across the ranked gene list. The peak of the curve is the enrichment score. Genes from the pathway are shown as vertical bars (the “rug”) at the bottom.
# Top activated pathway
top_up <- gsea_results %>%
as.data.frame() %>%
filter(NES > 0) %>%
slice_min(pval, n = 1) %>%
pull(pathway)
# Top suppressed pathway
top_down <- gsea_results %>%
as.data.frame() %>%
filter(NES < 0) %>%
slice_min(pval, n = 1) %>%
pull(pathway)
if (length(top_up) > 0) {
plotEnrichment(pathways_list[[top_up]], rank_vector) +
labs(title = paste("Top activated:", top_up))
}
if (length(top_down) > 0) {
plotEnrichment(pathways_list[[top_down]], rank_vector) +
labs(title = paste("Top suppressed:", top_down))
}